В основе этого довольно редкого заболевания лежит врожденная недостаточность остеобластической деятельности, которая сказывается в нарушении периостального остеогенеза при нормальном эпифизарном окостенении.
1. НЕСОВЕРШЕННЫЙ ОСТЕОГЕНЕЗ (OSTEOGENESIS IMPERFECTA)
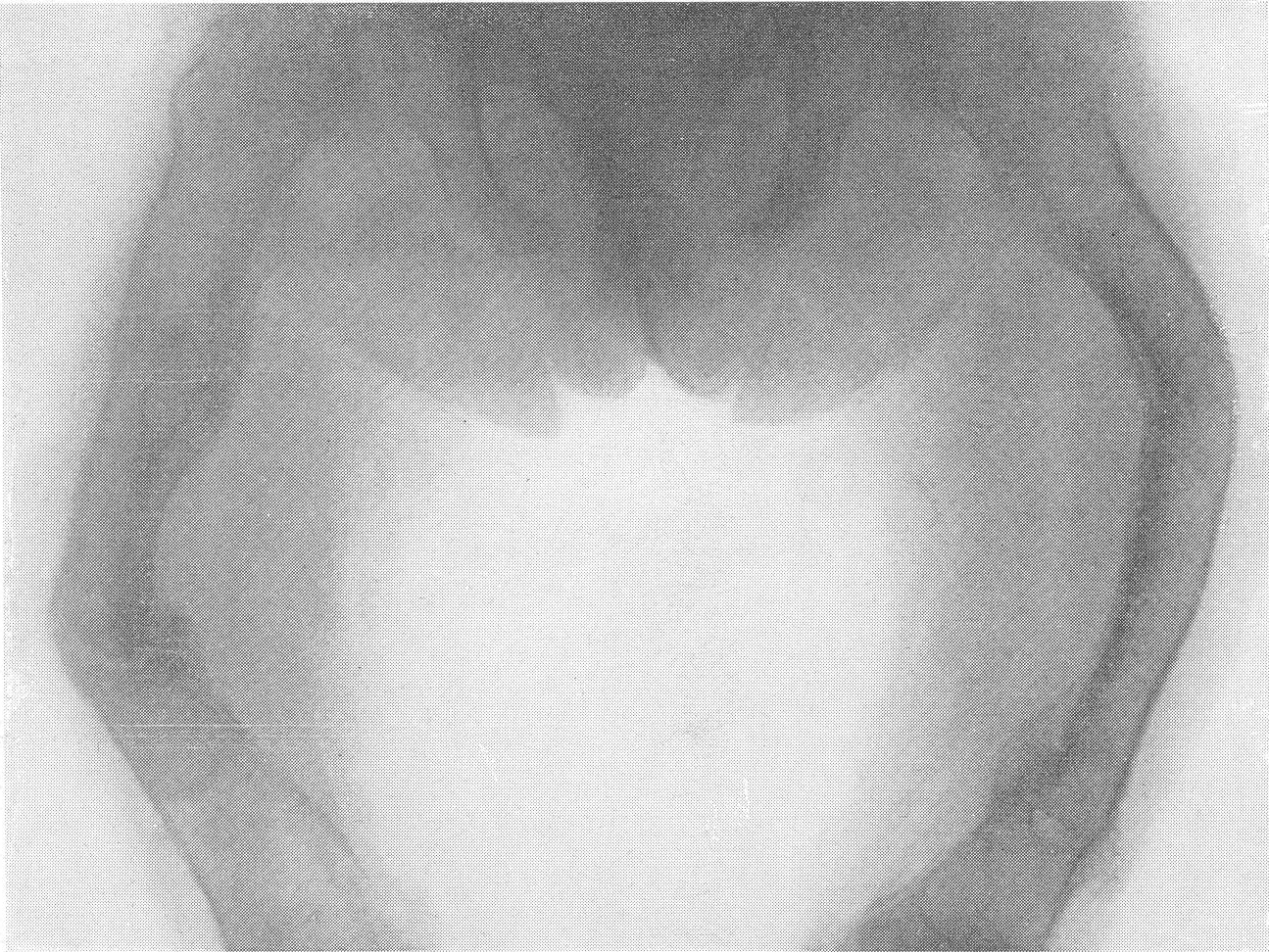
В основе этого довольно редкого заболевания лежит врожденная недостаточность остеобластической деятельности, которая сказывается в нарушении периостального остеогенеза при нормальном эпифизарном окостенении. Это своеобразный порок мезобластической дифференцировки и развития. Так как орган роста в длину — эпифизарный — особых патологических изменений не представляет, трубчатые цилиндрические кости нормальным образом растут в длину. В то же время рост в толщину вследствие понижения периостальной и эндостальной функции значительно подавлен и качественно изменен. Имеются указания на то, что наружный волокнистый (фиброзный) слой надкостницы утолщен. Внутренний же, камбиальный слой надкостницы вырабатывает собственно не обычные остеобласты, а скорее всего крупные хрящевые клетки. В тех местах, где вообще происходит обызвествление, оно совершается почти нормально. Остеокластическая деятельность не изменена, усиленного рассасывания кости не бывает. Вообще же кость при osteogenesis imperfecta очень бедна костными элементами. Развивающаяся костная ткань имеет не правильное пластинчатое строение, организованное вокруг гаверсовых каналов, а какой-то особенный зернистый или пористый вид. Многочисленные пазухи между костными островками заполнены рыхлой соединительной тканью. Корковое вещество весьма истончено. Все это вместе взятое и снижает механические качества кости, а следствием этого служит основное внешнее проявление заболевания, а именно — патологическая ломкость костей. Этиология несовершенного остеогенеза неясна. Поэтому, естественно, литература изобилует множеством деклараций и научных спекуляций, а хорошо обоснованных фактических данных, можно сказать, не имеется. Какие только предположения не высказаны за сто с лишним лет — с тех пор как Вролик (Vrolik) в 1845 г. впервые подробно описал эту болезнь у новорожденного ребенка и дал ей ныне общепринятое название osteogenesis imperfecta? Литература богата описаниями многих сотен семейств — носителей этой болезни. Довольно многочисленны сообщения о поражениях ряда поколений — трех, четырех, даже пяти поколений. Вообще семейный анамнез положителен в 10—25% всех случаев, в большинстве же случаев семейное влияние не прослежено и не доказано. Преимущественного поражения того или иного пола мы на нашем материале не наблюдали. Несовершенный остеогенез — это отнюдь не шаблонная болезнь, наоборот, клиническая и особенно рентгенологическая картина ее отличается значительным многообразием, которое определяется формой болезни, ее фазой, возрастом больного, степенью выраженности, особенностями лечения и другими условиями. Стремление выделить в рамках одной и той же нозологической единицы ряд и даже множество отдельных форм заболевания научного и практического смысла не имеет. Это ведь только индивидуальные особенности в каждом отдельном случае, отдельные сочетания симптомов, которые в лечебном отношении нас не вооружают. Действительно типичным является несовершенный остеогенез разве только у новорожденных и особенно у мертворожденных. Эту форму — внутриутробную (фетальную), или болезнь Вролика, с полным основанием следует противопоставить всем другим проявлениям этой болезни. Кроме внутриутробной, принято различать детскую форму, юношескую и форму, поражающую людей после периода полового созревания, т. е. взрослых. Формы болезни, которые появляются впервые после рождения ребенка, в момент рождения видимо внешне здорового, объединяются названием osteogenesis imperfecta tarda, fragilitas ossium, или идиопатическая ломкость костей— osteopsathyrosis idiopathica. Если некоторые авторы в свое время считали остеопсатироз за совершенно самостоятельное заболевание, а именно вовсе не врожденное, а приобретенное — за авитаминоз, не имеющий по своему существу ничего общего с несовершенным остеогенезом, то теперь нет никаких сомнений в том, что это груб\’о ошибочная точка зрения. Наибольший процент случаев падает при несовершенном остеогенезе на внутриутробную форму, при которой на свет появляются чаще всего мертворожденные. Живорожденные очень слабы, дают стойкие и длительные профузные поты, петехиальные кровоизлияния, синюху, нередко умеренные повышения температуры, и болезнь отличается, несмотря на все меры профилактики, исключительно высокой смертностью в первые же дни и недели, редко — месяцы жизни. Чем старше ребенок, тем реже встречается болезнь. Чем раньше она проявляется, тем хуже предсказание в смысле жизни. И тем не менее противопоставление внутриутробной форме несовершенного остеогенеза идиопатического остеопсатироза глубоко обосновано именно по той причине, что последний вполне совместим с жизнью и приобретает живой клинический интерес. В отдельных случаях больные идиопатическим остеопсатирозом достигают зрелого и даже пожилого возраста, например, шестого и седьмого десятилетия жизни в серии наших собственных наблюдений. Как правило, они при этом отличаются нормальным умственным развитием. Что же касается степени инвалидизации выживающих, то она колеблется в весьма широких пределах, и мы наблюдали как лиц с почти сохраненной работоспособностью, так и полукалек, передвигающихся на тележках, и полных калек, прикованных годами к кровати и совершенно беспомощных. Будучи во внешней картине болезни ведущими, костные изменения не являются, однако, единственными. Уже с 80-х годов прошлого столетия известно, что несовершенный остеогенез сочетается с другими характерными болезненными явлениями. На первом месте стоят так называемые синие склеры. Это ни с чем не смешиваемая своеобразная окраска белковой (и роговой) оболочки глаз — синяя, голубая, подчас с серым отливом. Объясняется этот симптом тем, что склеры и роговицы будто бы истончены примерно до 1/3 своей нормальной толщины. Имеются также указания на весьма глубокие качественные изменения этих мезенхимальных по происхождению оболочек глаза. Как бы то ни было, склера является тонкой, мягкой, податливой — лептосклерия, и лежащая под ней сосудистая сеть просвечивает, этим самым меняя окраску белков глаз и производя на врача странное впечатление, пока оно не осознано. Лептосклерия в более или менее резко выраженной форме всегда налицо, если только имеются костные проявления несовершенного остеогенеза. Синие склеры могут к тому же быть в пораженных семьях у других членов семьи, даже и вовсе не страдающих костными поражениями. Вот почему синим склерам по справедливости придается очень большое дифференциально-диагностическое значение. Гораздо менее известна голубая окраска барабанных перепонок, которая, естественно, не так обращает на себя внимание и требует специальных методов определения. Характерным общим симптомом несовершенного остеогенеза служит и отосклероз. Тугоухость и глухота не наблюдаются до периода полового созревания, а падение слуха развивается, прогрессивно нарастая с различной быстротой, лишь начиная с возраста 20—30 лет. Почти общее мнение таково, что причина отосклероза при этом заболевании обычная, т. е. заключается в нарастающем фиброзном, а затем и костном анкилозе суставчиков между мелкими косточками в полости среднего уха (молоточком, наковальней и стремечком). Отосклероз встречается реже лептосклерии и, значит, не может считаться постоянным симптомом болезни. Тем не менее принято говорить о часто наблюдаемой и характерной триаде — ломкости костей, синих склерах и отосклерозе. Следует также указать на мало известную, но довольно частую при несовершенном остеогенезе коричневатую окраску зубов, объясняемую особого вида прозрачностью зубной эмали. В этой связи стоят также встречающиеся уже гораздо более редко неправильности развития зубов, ногтей на пальцах рук и ног, а также особенности волос, притом в самых различных сочетаниях. Наконец, несовершенному остеогенезу как болезни свойственны слабость, какая-то дряблость, недоразвитость суставных сумок и всего связочного суставного аппарата. В результате этого больные имеют, помимо костных изменений, еще разболтанные суставы; сравнительно часто наступают разрывы связок и даже вывихи. Один из наших больных использовал этот свой врожденный повышенный объем подвижности в суставах и в позвоночном столбе, к тому же доведенный путем упражнения с ранних детских лет до виртуозной крайности, для работы в цирке в качестве „резинового человека”. Подчеркнем, что биохимическое исследование крови при несовершенном остеогенезе в общем сдвигов не выявляет. Как правило, содержание кальция и фосфора в крови находится на нормальном уровне. Возможно, что временные и незначительные колебания этих показателей минерального баланса совпадают с фазами обострения заболевания и учащением переломов. Что же касается фосфатазы сыворотки крови, то по этому важному вопросу имеются резкие разногласия, и он далеко еще не разрешен. Биохимические данные имеют немаловажное практическое значение в случаях сочетания нескольких заболеваний, а также в отличительном распознавании. Эндокринные железы при несовершенном остеогенезе оказываются по всем клиническим, рентгенологическим, а также весьма многочисленным секционным данным неизменно непричастными, сама по себе эта врожденная болезнь этиологически и патогенетически с железами внутренней секреции не связана. Что касается нервной системы, центральной и периферической, то, к сожалению, этот вопрос остается в свете современных повышенных методических требований неизученным. Рентгенограммы точно передают основные грубо морфологические и функциональные особенности картины патологической перестройки при несовершенном остеогенезе, и мы рассмотрим патологоанатомическую и рентгенологическую картины этой болезни вместе. Кости соответственно ничтожному содержанию костных элементов и минеральных солей представляются при внутриутробной фор ме болезни рентгенологически необычайно прозрачными. Корковый слой может быть истончен до толщины бумаги, а местами, где надкостница в пределах диафиза непосредственно прилегает к губчатому веществу, компактная краевая каемка рентгенологически и вовсе отсутствует. Костномозговой канал в связи с этим эксцентрически увеличен в диаметре и местами неровен. Губчатая структура разрежена и имеет широкопетлистый, сетчатый, а иногда неправильный хаотический рисунок, отдельные трабекулы и на прекрасных в техническом отношении снимках иногда едва выступают. В тяжелых случаях костная ткань настолько поротична, что по своей нежной гомогенной тени ничем не отличается от окружающих мягких тканей, так что кость может быть узнана лишь благодаря тонким „волосяным” линиям коркового слоя. Особенно характерно множество переломов больших трубчатых костей. Прозрачный скелет становится хрупким как стекло; отсюда название „стеклянные люди”. Переломы чаще всего поражают средние отделы диафизов бедра и плеча, затем костей голени и предплечья, часто на симметричных местах. Особенно много переломов видно также по ходу ребер. Эти переломы являются чаще всего полными поперечными, реже частичными, в виде под-надкостничных надломов. Очень большого смещения отломков из-за слабости мышц не приходится наблюдать. Более значительным и характерным является угловое смещение отломков в средней трети бедра и плеча, причем вершина угла смещения направлена вверх и в латеральную сторону, иногда видно и небольшое боковое смещение. Множественные переломы больших трубчатых костей ведут к значительному укорочению и деформации костей, и пораженные кости приобретают внешнее сходство с гармоникой (рис. 261, Б). Деформации и главным образом утолщению костей способствуют старые и более свежие костные мозоли. Несмотря на ничтожную функциональную ценность надкостницы, а может быть, и в результате обширных кровоизлияний эти костные мозоли очень быстро появляются и достигают больших размеров. Они обыкновенно имеют шаровидную форму и симметрично охватывают место перелома. Значительное обызвествление мозоли не наблюдается. На месте перелома появляются более плотные костные трабекулы. По степени обызвествления и по относительной интенсивности губчатого рисунка на концах отломков можно судить о давности перелома.
{kind=link}
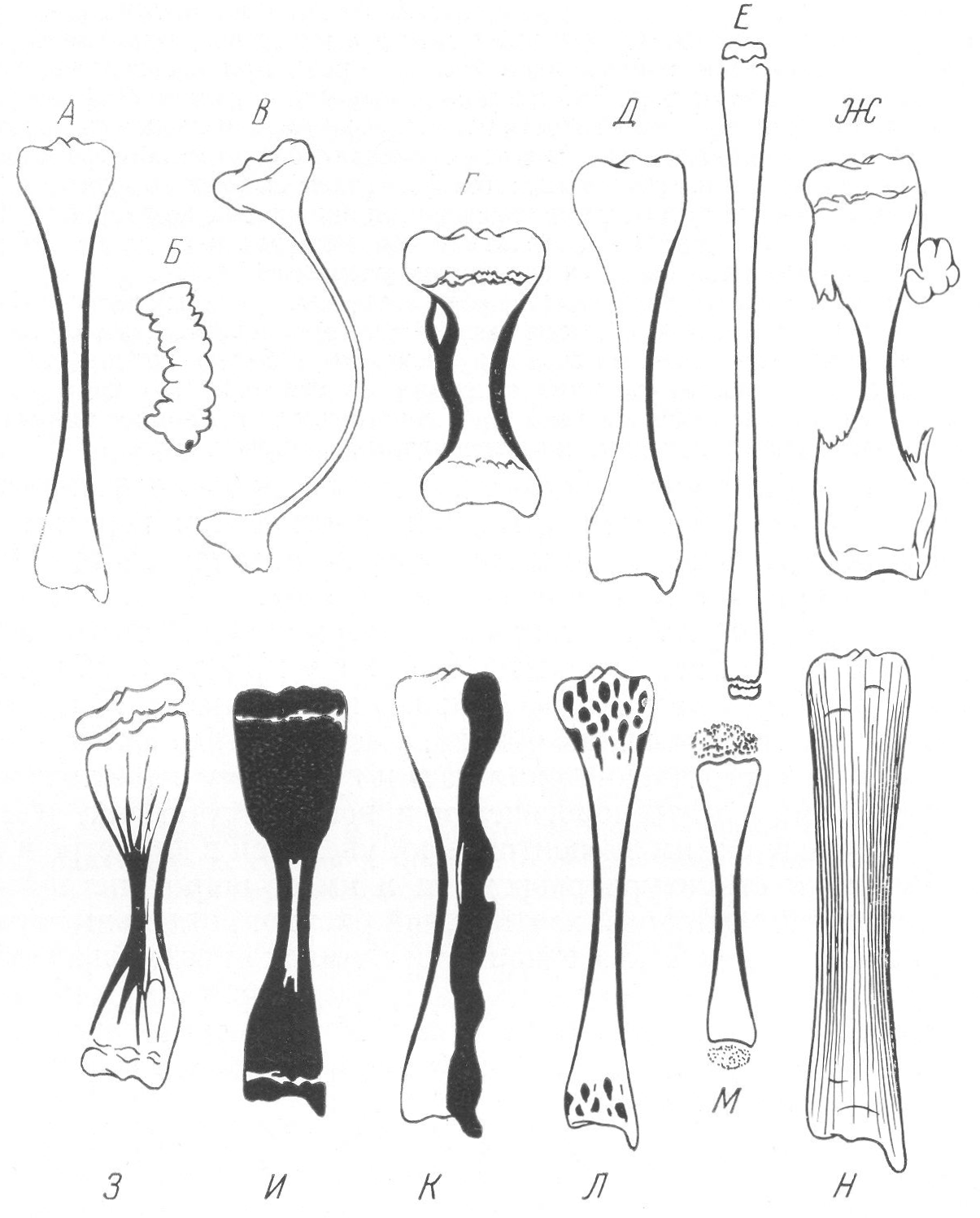
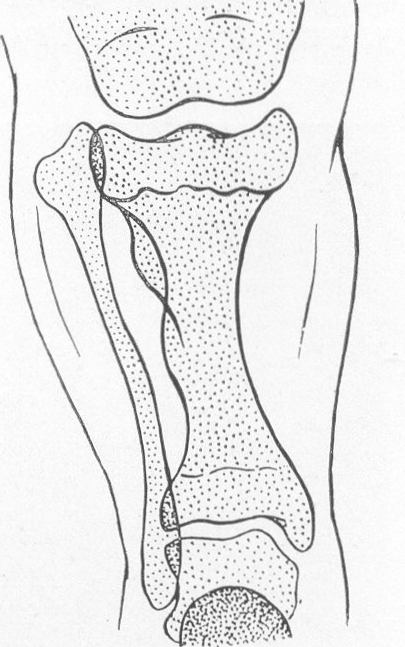
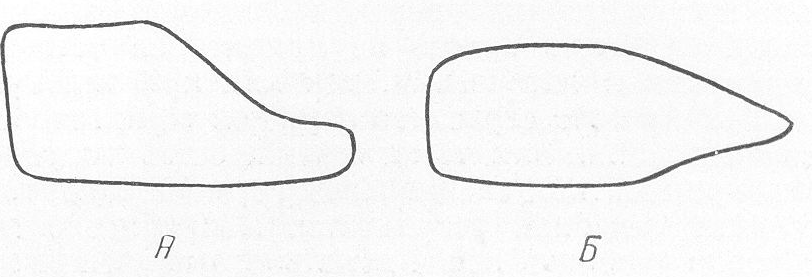
Рис. 261. Схематическое изображение основных рентгенологических проявлений деформации длинных трубчатых костей при различных врожденных системных заболеваниях скелета. А — нормальная большеберцовая кость для сравнения; Б — несовершенное костеобразование; В — идиопатический остеопсатироз; Г — хондродистрофия; Д — остеохондродистрофия; Е — арахнодактилия; Ж — множественные хрящевые экзостозы; 3 — множественный хондроматоз костей; И — мраморная болезнь; К — мелореостоз; Л — остеопойкилия; М — врожденная эпифизарная точечная дисплазия; Н — врожденные системные диафизарные гиперостозы.
Если переломы расположены близко друг от друга, например если одновременно имеется 5—8 и больше переломов диафиза, то мозоли различной давности и величины сливаются вместе, и кость приобретает своеобразный неуклюжий узловатый обезображенный вид. По этой причине подчас наблюдается до известной степени парадоксальная картина, т. е. не крайнее истончение коркового слоя кости, а некоторое, хотя бы частичное его утолщение. В результате бесчисленных переломов и надломов с последующим образованием периостальных мозолей и вдвиганием наподобие подзорной трубы (телескопическое укорочение длинной трубчатой кости) корковый слой, действительно, может оказаться утолщенным и костномозговой канал суженным и эксцентрически смещенным.
{kind=link}
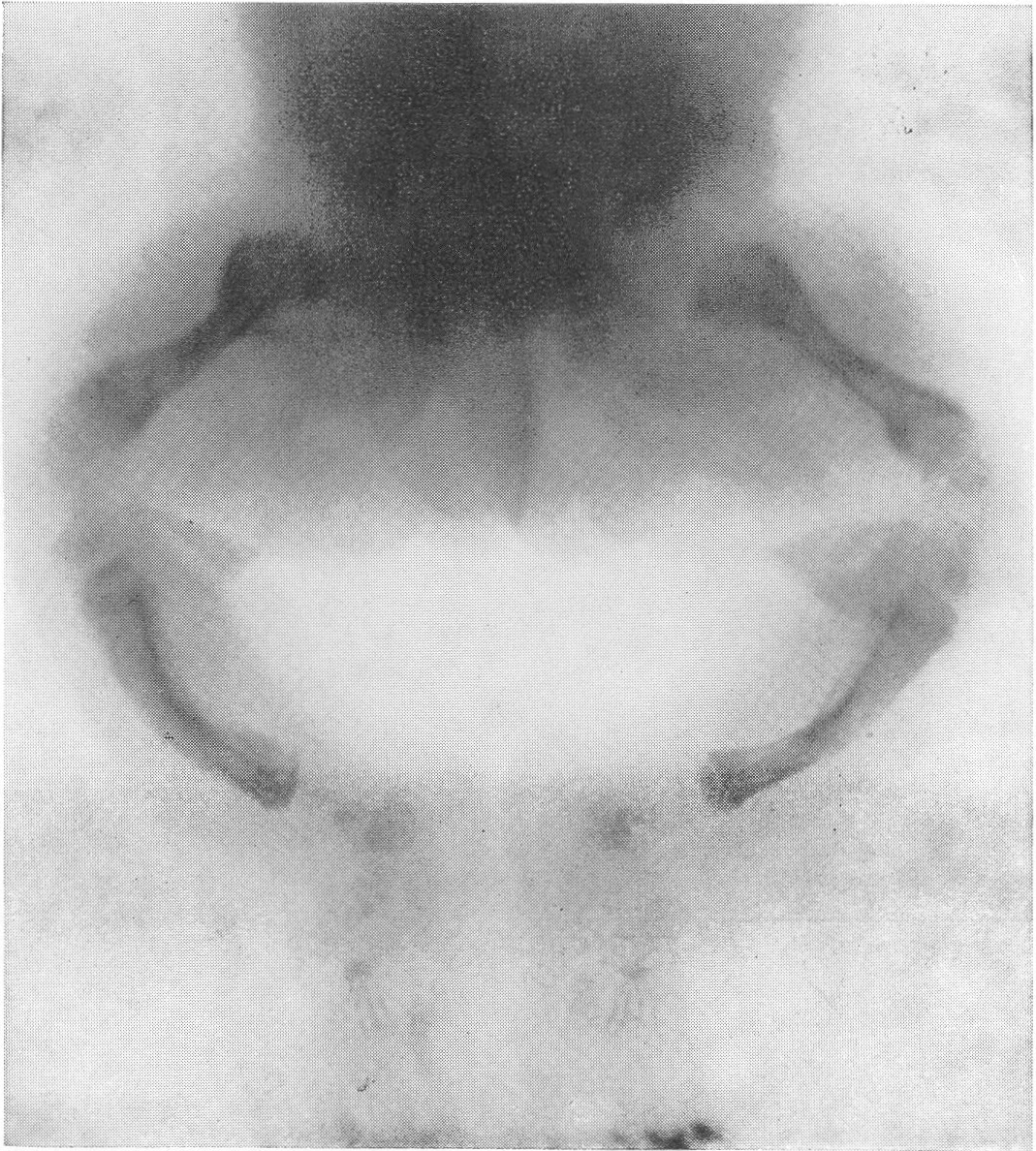
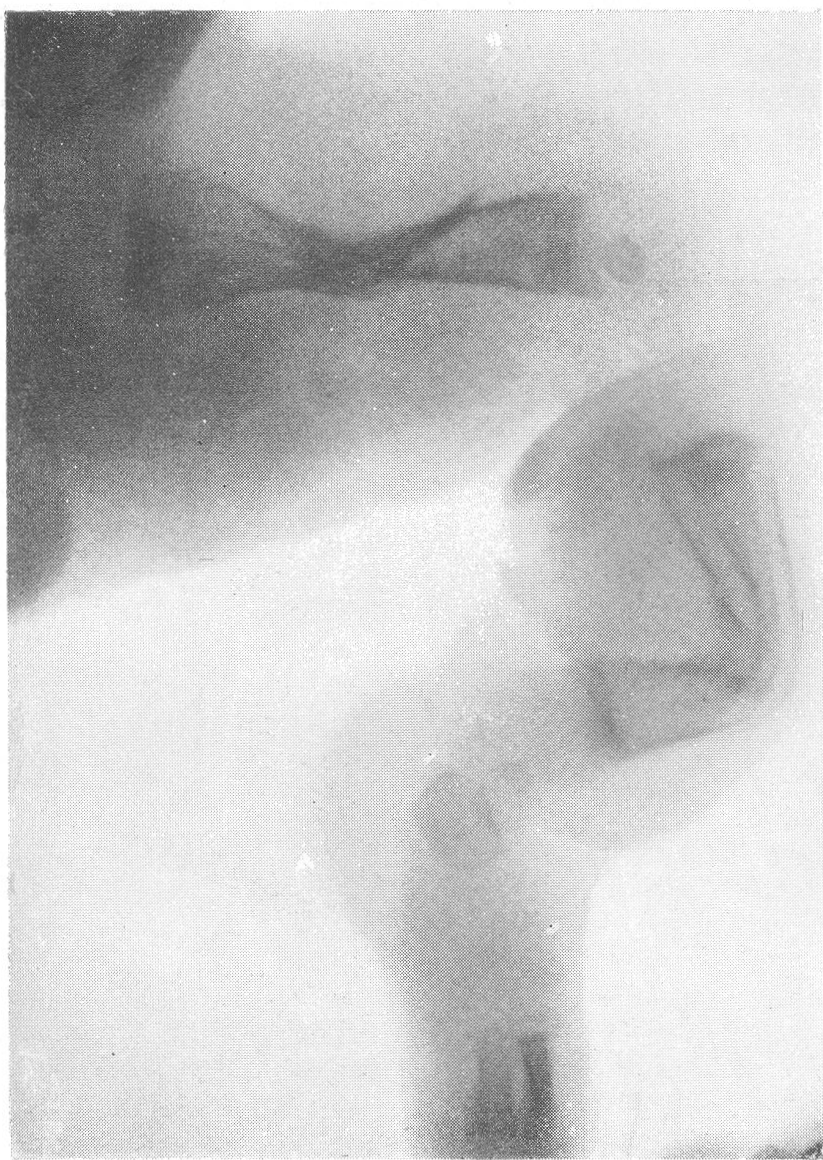
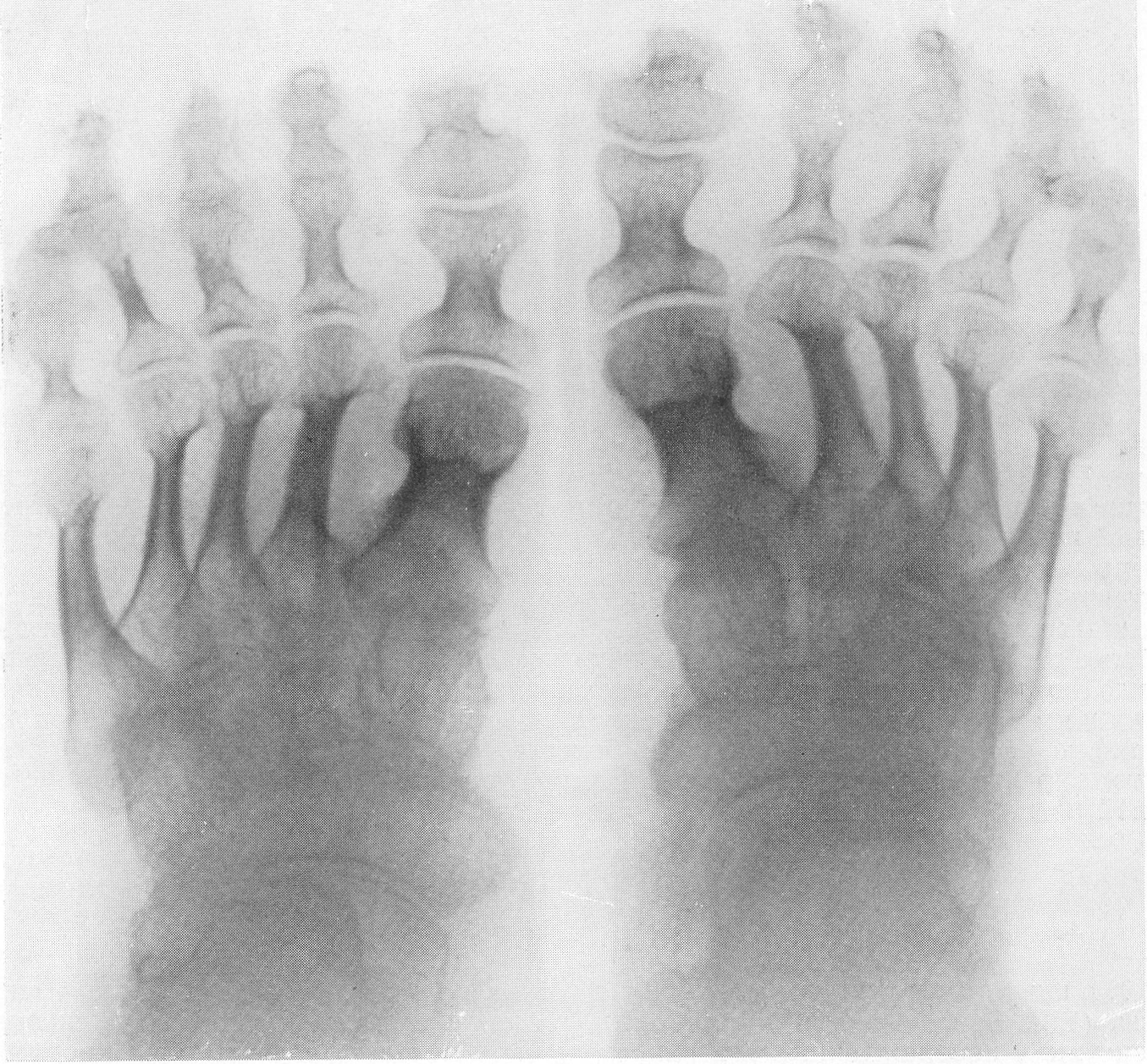
Типичная рентгенологическая картина нижних конечностей при множественных переломах. Это наблюдается главным образом на тех местах конечностей, которые поддерживаются одной большой трубчатой костью, т. е. на бедре и на плече, но не на голени и на предплечье. Укорочение кости при внутриутробной форме несовершенного остеогенеза зависит только от переломов, и чем больше их число, тем больше укорачивается кость. Общее количество переломов всех костей может равняться многим десяткам, а в редких казуистических случаях доходить и до нескольких сотен. Чем больше число переломов, особенно свежих, тем хуже предсказание. А так как рентгенологически неожиданно для клинициста обнаруживается всегда больше переломов, чем это предполагалось, и только рентгенограммы дают возможность с точностью сосчитать их число и следить за их новым появлением, то рентгенологическое исследование имеет и большое прогностическое значение. Вследствие поражения главным образом длинных периферических костей конечности становятся непропорционально короткими, и при нормальных размерах головы и туловища развивается микромелия (рис. 262 и 263). Рентгенологически это выражается, как и клинически, и в том, что кожа, покрывающая укоротившиеся кости, ложится в поперечные складки. Эти складки видны на хороших рентгенограммах и неопытным исследователем могут быть приняты на снимках за линии перелома; от переломов, как известно, поперечные кожные складки отличаются тем, что лучше всего видны на поверхности контуров мягких тканей и в их тени, т. е. и вне тени костного цилиндра. Очень характерным признаком osteogenesis imperfecta являются нормальные размеры кистей и стоп — фаланги никогда не ломаются, а плюсневые и пястные кости — лишь в редчайших случаях, и то не все, а только одна или две кости.
{kind=link}
{kind=link}
Поэтому пальчики при коротких неуклюжих толстых более проксимальных отделах кажутся несоразмерно длинными и заостренными. Никогда при врожденном заболевании не ломаются основание черепа, позвоночник и грудина.
{kind=link}
Рис. 265. Рентгенограмма нижних конечностей при несовершенном костеобразовании у грудного ребенка с ограниченным числом переломов бедер и костей голени.
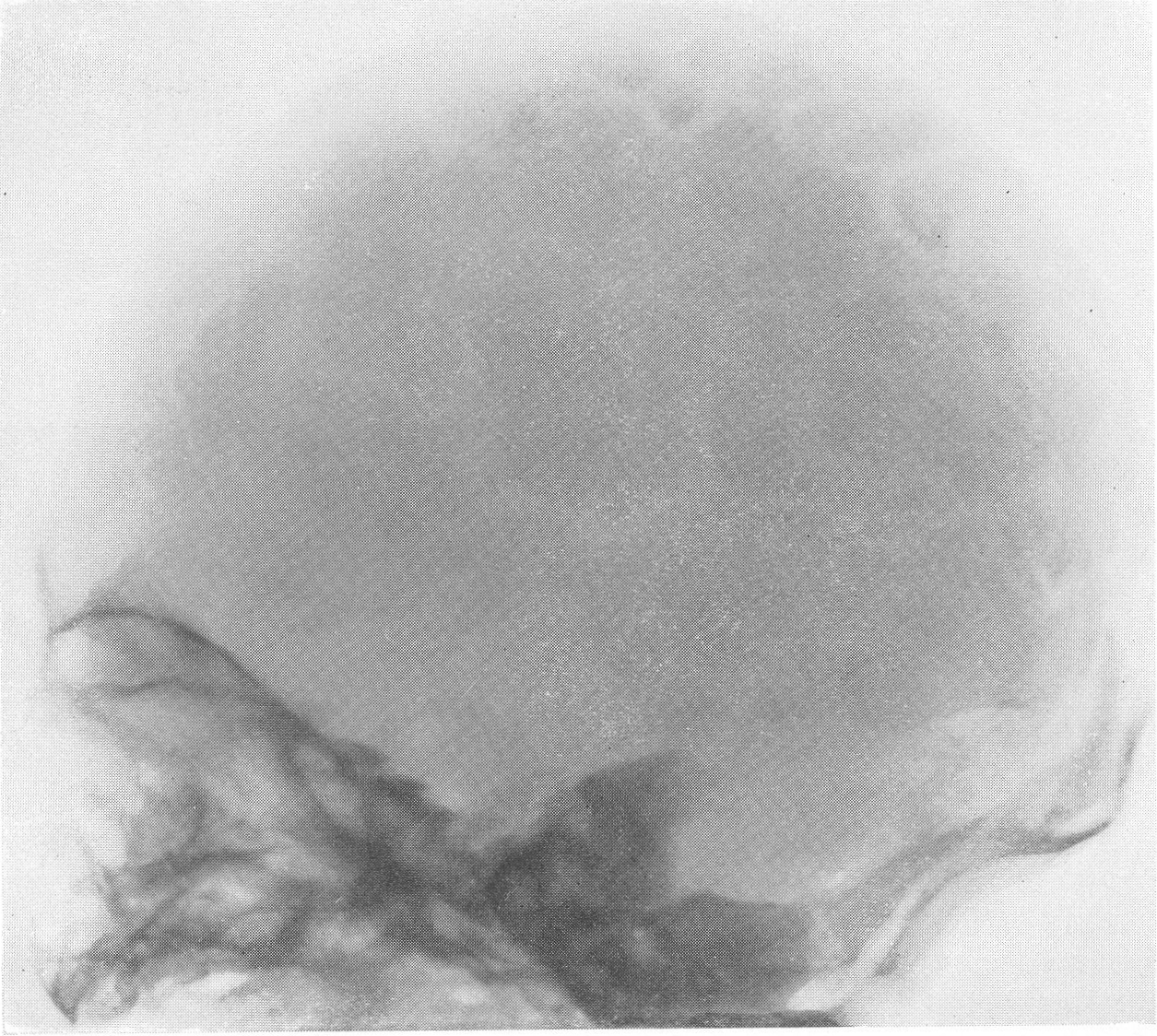
Эпифизарные полоски несколько разрыхлены или же особых изменений не представляют. Эпифизарные ядра окостенения появляются в срок и также обрисовываются вполне нормально, если не считать общего для всего скелета остеопороза. Очень характерную рентгенологическую картину представляет череп (рис. 264), клинически при пальпации напоминающий мягкий, пластический, податливый кожистый мешок. На фоне мягкой тени свода черепа обрисовываются отдельные крапинки, или лишь единичные мозаичные, ландкартообразные, или округлые звездчатые, с бахромчатыми неровными краями островки обызвествления, и эти тонкие пластинки то связаны, то не связаны друг с другом. При тангенциальном ходе лучей выясняется, что покровные кости черепа необычайно истончены. Затылочная кость, в особенности ее основание, обыкновенно лучше обызвестлена. По форме череп ребенка при несовершенном остеогенезе не похож на гидроцефалический и тем более на хондродистрофический череп. В частности, носик ребенка имеет нормальную спинку, не запавшую, как это свойственно хондродистрофии. Детская форма несовершенного остеогенеза, появляющаяся при первых попытках встать на ножки или ходить, в возрасте до 1 1/2—2 лет, обозначаемая также как болезнь Лобштейна (Lobstein), по имени видного в середине прошлого века французского патологоанатома, а также поздняя форма — это более легкие случаи врожденного заболевания.
{kind=link}
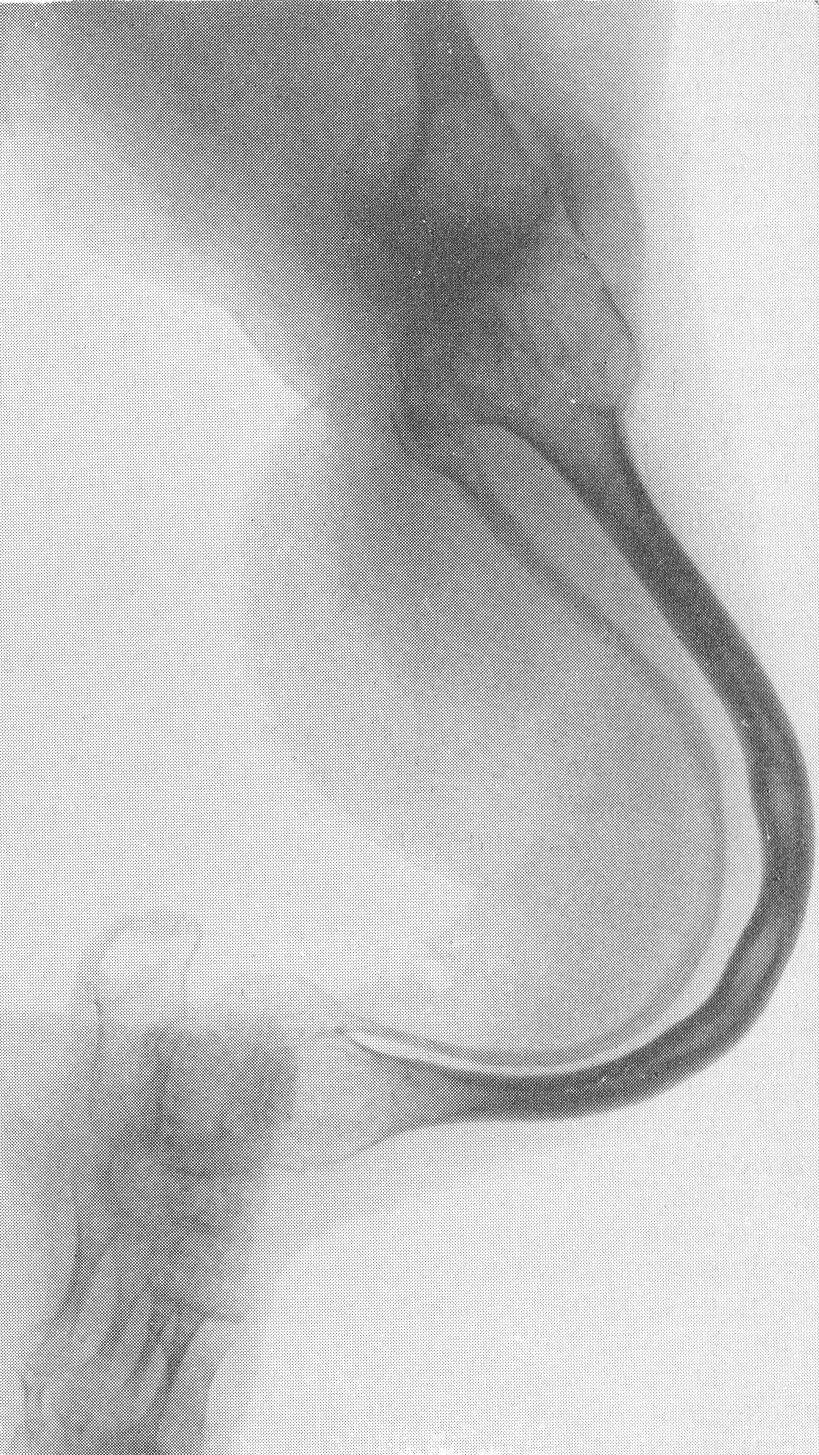
Рентгенологическая картина этого так называемого идиопатического остеопсатироза несколько отличается от только что описанной (рис. 265 и 266). Прежде всего количество переломов никогда не бывает так велико, как при явной врожденной форме. Обычно приходится видеть одновременно всего 3—6 переломов костей. Чаще всего ломаются кости нижней конечности, в особенности бедро, реже кости голени. Длинные трубчатые кости не укорочены и не утолщены, а, наоборот, кажутся очень стройными и грацильными (рис. 261, В). Остеопороз на нижних конечностях, еще и вследствие недеятельности, резче выражен, чем на верхних, так что, например, необычайно тоненькая малоберцовая кость едва выделяется на снимке. Эпифизарные костные концы кажутся относительно толстыми. Бедро дугообразно искривлено вверх и наружу, как при рахите, и сохраняет нормальную длину, большеберцовая кость имеет вид сабельных ножен, выступающих острым передним гребнем вперед (рис. 267). Изменения обычно симметричны, но с обеих сторон не строго зеркальные. На месте перелома после его заживления деформация подчас остается не дугообразной, а резко угловой, аналогично не кифозу позвоночника, а угловому горбу. Это так называемая ангуляция конечности. Очень часто видны лоозеровские зоны, которые не следует смешивать с истинными переломами. Костные мозоли не имеют таких больших размеров, зато лучше обызвествлены. У детей старшего возраста иногда наблюдается довольно сложная по своему структурному рисунку перестройка костной ткани. В очень редких случаях кость может приобрести крупноячеистый кистозный вид, так что некоторые авторы говорят об особенной кистовидной рентгенологической форме несовершенного остеогенеза — об osteogenesis imperfecta cystica (Fairbank).
{kind=link}
Сходство это не только внешне формально, но, вероятно, обусловлено после многократных переломов значительными кровоизлияниями и последовательными восстановительными явлениями. Эта разновидность наблюдается почти исключительно в одном излюбленном месте — в дистальных двух третях бедренной кости. Позвоночник, который в общей картине интересующего нас здесь заболевания вообще-то принимает малое участие, во всяком случае при внутриутробной форме, у выживающих больных все же также может быть вовлеченным в патологический процесс. Все позвонки структурно изменены, поротичны, прозрачны, тела слегка расширены. Иногда на всех, на многих или на отдельных телах площадки сильно вдавлены — тела двояковогнуты, а межпозвонковые диски, наоборот, высоки, чечевицеподобны, двояковыпуклы. Поэтому не удивительно, что в редких случаях, когда годами не прекращаются тяжелые активные поражения как конечностей, так и позвоночника, наступают процессы отставания роста и даже уменьшение достигнутого роста, словом, та или иная степень карликовости. Довольно характерную картину представляет при остеопсатирозе и череп. Широкий лоб выпячивается вперед и круто поднимается, височные кости нависают над наружными слуховыми проходами, а уши оттопырены книзу и кнаружи. Весь мозговой череп велик и производит впечатление неправильного усеченного конуса, вершиной покоящегося на позвоночнике. Иногда и у взрослых остаются зияющими швы, они даже могут быть прощупываемы. Кости свода крайне истончены. Как правило, видны многочисленные добавочные вормиевы вставочные косточки швов, ossa suturarum, так называемая ossa Wormiana. Имеются указания, что при общем остеопорозе массив каменистой кости не только не теряет костного вещества, но даже может склерозироваться, во всяком случае этот участок основания черепа с обеих сторон контрастно выступает на фоне более прозрачных, чем в норме, элементов черепа. Рентгенограммы имеют при несовершенном остеогенезе чрезвычайно высокое, можно сказать, почти патогномоничное диагностическое значение, так что в типичных случаях дифференциальной рентгенодиагностики собственно и не существует. Клинический же диагноз не всегда легок. Переломы наступают при одной только мышечной тракции и при ничтожных травмах во время манипуляций при родах или при пеленании ребенка. По-видимому, переломы не болезненные — через несколько дней после перелома ребенок уже свободно владеет сломанной конечностью. Поэтому естественно, что с клинической стороны в ряде случаев переломы и не предполагаются и обнаруживаются только на снимках. Обыкновенно думают о более известной и чаще встречающейся хондродистрофии, — ведь оба заболевания характеризуются микромелией. Одного взгляда на ручку или на снимок кисти, однако, достаточно, чтобы провести дифференциальный диагноз — нормально-пропорциональная кость при несовершенном остеогенезе резко отличается от типичной картины хондродистрофии, уже не говоря о других отличительных признаках (стр. 403). Рахит в соответствующем для него возрасте исключается на основании неизмененных эпифизарных зон при несовершенном остеогенезе, именно в ростковых хрящах при рахите разыгрываются главные патологические изменения. Легче всего отбросить врожденный сифилис. В одном из случаев автора несовершенный остеогенез шел до рентгенологического исследования под диагнозом множественных псевдопараличей Парро. О дифференциальной рентгенодиагностике между несовершенным остеогенезом и детской остеомаляцией см. кн. 2, стр. 30. В последние годы стало известно одно крайне интересное в теоретическом отношении и особенно важное в практическом дифференциально-диагностическом смысле, осложнение при позднем несовершенном остеогенезе. Речь идет о так называемых псевдосаркомах при osteogenesis imperfecta — о таких переломах, которые сопровождаются чрезвычайно обширными, чрезмерно массивными костными мозолями, симулирующими злокачественное новообразование. В литературе имеется всего около двух десятков подобных случаев. Дело в том, что и при обычных условиях костная мозоль при этом заболевании велика, но в процессе заживления кости эта мозоль претерпевает обратное развитие. В редких же случаях возникает, можно сказать, чудовищная костная мозоль, распространяющаяся подчас вдоль всего длинника диафиза кости, но главным образом возвышающаяся в стороны, над поверхностью кости. При этом костные разрастания весьма плотны, могут иметь поперечную исчерченность наподобие солнечных лучей или частокола. Клинически процесс бурного роста затвердения вокруг пораженной кости сопровождается чаще всего резкими самопроизвольными болями и чувствительностью при пальпации, напряжением и покраснением кожи, местным жаром и даже общим повышением температуры тела. Мы сами наблюдали один подобный случай у взрослого больного с тяжелым и далеко зашедшим поздним несовершенным остеогенезом (рис. 268). Опухолевидные массы появились неожиданно вокруг типично пораженного диафиза плечевой кости. От диагностической ошибки нас спасло лишь настойчивое указание умственно хорошо развитого глубоко инвалидизированного больного на то, что это повторное явление — в других длинных трубчатых костях (бедренной, другой плечевой) несколько лет назад аналогичные явления самостоятельно кончились благополучно. Фербенк и Бейкер (Fairbank a. Baker) в двух хорошо прослеженных случаях доказали, что гистологически никаких элементов злокачественной опухоли не было, плотные разрастания состояли из бурно пролиферировавшей соединительной ткани, содержавшей много волокнисто-слизистой (фибромукоидной) и хрящеподобной массы. Удивительно, что подобные местные опухолеподобные разрастающиеся костные массы могут возникать при несовершенном остеогенезе и самопроизвольно без всякого перелома, вокруг ненарушенной в целости кости. При этом настоящий перелом в той же кости или в других местах скелета заживал ранее обычным образом, т. е. без чрезмерного образования мозоли. Поэтому нет основания приписывать эту из рук вон выходящую реакцию кровоизлияниям, повреждениям мышц. Особенно же следует отвергнуть допущение о присоединившейся цинге, как это считает Брейлсфорд (Brailsford). Это важно учесть по той причине, что препараты витамина С при этом никакого положительного действия не оказывают. Природа этого осложнения при несовершенном остеогенезе остается совершенно невыясненной и загадочной. При основном заболевании, сущность которого заключается в слабости, в неактивности периостальной остеобластической деятельности, мы неожиданно наблюдаем явно противоположные качества, лежащую на противоположном полюсе крайность — беспрецедентную в костной патологии надкостничную остеобластическую сверхактивность! Принципиально важно особенно подчеркнуть, что формальная клиническая и рентгенологическая картина на высоте заболевания говорит в пользу характернейшей остеогенной саркомы.
{kind=link}
Огромные опухолевидные костные разрастания вокруг плечевой кости с последующим инволютивным течением и постепенным рассасыванием. Действительно, самые опытные исследователи неминуемо совершат опасную диагностическую ошибку, если не будут знакомы с этим пусть и весьма редким осложнением редкого заболевания. Это один из ярких примеров псевдосарком. И при всем сказанном не следует еще упускать из виду то обстоятельство, что при несовершенном остеогенезе в качестве редчайшей казуистики все-таки возможно и истинное озлокачествление. Правда, в свете точных современных наблюдений о псевдосаркомах при несовершенном остеогенезе эти старые указания о малигнизации вызывают обоснованное сомнение. О лечении несовершенного остеогенеза можно написать коротко — все только мыслимое испробовано и притом до сих пор неизменно безуспешно. Всевозможные эндокринные, гормональные, витаминные, диететические и другие препараты оказались без заметного лечебного влияния. В частности, беспомощны все препараты, производные околощитовидной железы, при помощи которых, казалось бы, следовало пытаться повлиять на минеральный обмен. Учитывая правильное старое наблюдение об улучшении состояния больных с их вступлением в период полового созревания, стали испытывать половые гормональные продукты. Однако определенного положительного впечатления от их действия тоже не получается. Остаются одни только общие щадящие мероприятия, призванные предотвратить или по крайней мере сократить число неминуемо наступающих переломов и дальнейшее калечение несчастных больных. Сами же переломы поддаются обычному лечению вполне успешно, обычно даже в сокращенные сроки.
2. ХОНДРОДИСТРОФИЯ
Это довольно редко встречающееся врожденное заболевание известно с древних времен. Оно в течение ряда предыдущих столетий оставило более глубокий след в искусстве, чем в науке. Достаточно указать на непревзойденные по мастерству изображения хондродистрофических карликов, которых так охотно рисовал великий испанский живописец XVII века Веласкез. Название этой болезни, теперь общепринятое у нас, дано Кауфманом (Kaufmann) в 1892 г. Еще в 1876 г. Парро (Parrot), а в 1900 г. Пьер Мари (Pierre Marie) представили очень подробное научное описание этой столь волновавшей воображение человечества в течение веков болезни. Отсюда — „болезнь Парро—Мари”. Французское название — ахондроплазия следует считать неточным и явно неудачным, но знать и этот синоним необходимо, так как в литературе и это обозначение широко привилось и до сих пор еще многими учеными и врачами применяется. Другой синоним — микромелия („малые члены”, мелос по-древнегречески — член) является слишком расплывчатым и общим для многих заболеваний, а поэтому смысла не имеет. В основе хондродистрофии лежит нарушение энхондрального развития костного скелета: поражаются именно только те кости, которые растут по энхондральному типу, кости же, обызвествляющиеся по соединительнотканному пути, как, например, в первую очередь покровные кости черепного свода и ключица, патологических изменений не представляют, или же поражаются в незначительной степени лишь в очень тяжелых случаях chondrodystrophia foetalis. По своему существу хондродистрофия представляет собой прямую противоположность несовершенному остеогенезу. При osteogenesis imperfecta нарушено периостальное и эндостальное окостенение при неизмененном энхондральном процессе, при хондродистрофии же, наоборот, периостальный и эндостальный рост в толщину протекает нормально, а рост в длину вследствие неправильного энхондрального окостенения, т.е. преимущественно в эпифизарных концах длинных костей, подавлен. Главнейшим проявлением хондродистрофии служит укорочение пораженных костей. Кауфман, а за ним и большинство авторов различают три формы хондродистрофии: chondrodystrophia hyperplastica, hypoplastica и malacica. Всем формам гистологически свойственно беспорядочное анархическое расположение хрящевых клеток в ростковом хряще в отличие от нормальной правильной картины, когда хрящевые клетки расположены ровными столбами. Общим признаком всех трех форм является неправильный способ обызвествления с нарушением роста кости. Гиперпластическая форма при этом характеризуется чрезмерным разрастанием и увеличением размеров эпифизарного хряща, гипопластическая, наоборот, уменьшением; третья форма отличается размягчением хряща. Chondrodystrophia malacica, наиболее редко встречающаяся, не имеет практического значения, так как она несовместима с жизнью, на свет появляются недоношенные мертвые плоды. Гипер- и гипопластическая формы могут быть дифференцированы в последние месяцы эмбрионального развития, в дальнейшем же они недостаточно надежно отличаются друг от друга. С практической точки зрения распознавание формы и не требуется. И при этих формах нередко наблюдаются мертворожденные, но выживающие хондродистрофики очень жизнеспособны и могут доживать до глубокой старости. Умственное развитие происходит нормально. Каких-нибудь серьезных изменений нервной, а также эндокринной системы, как и других систем организма, кроме костной, никогда никто не находил. Женский пол заболевает немного чаще мужского. Изменения костей при хондродистрофии имеют строго зеркальный симметричный характер. В первую очередь поражаются кости конечностей, причем более проксимальные в большей степени изменены, чем дистальные: бедра и плечи резче всего укорочены, кости голени и предплечья — в меньшей степени и еще меньше — плюсневые, пястные кости и фаланги. Во вторую очередь в патологический процесс вовлекаются ребра, тазовые кости и основание черепа, и меньше всего изменены позвоночник и остальные кости туловища, особенно ключица. Хондродистрофия, таким образом, ведет к карликовому росту и является наиболее частой причиной карликовости.
{kind=link}
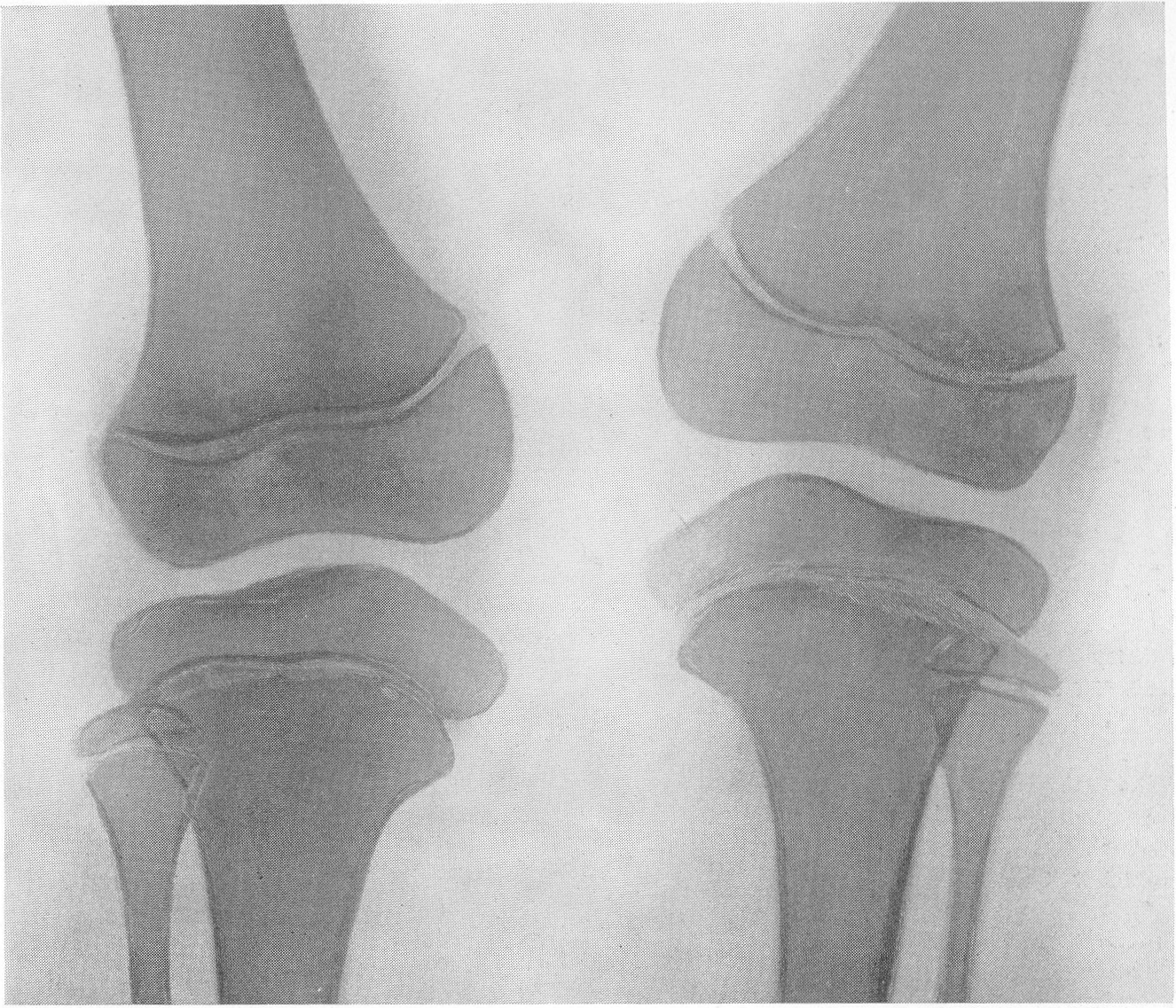
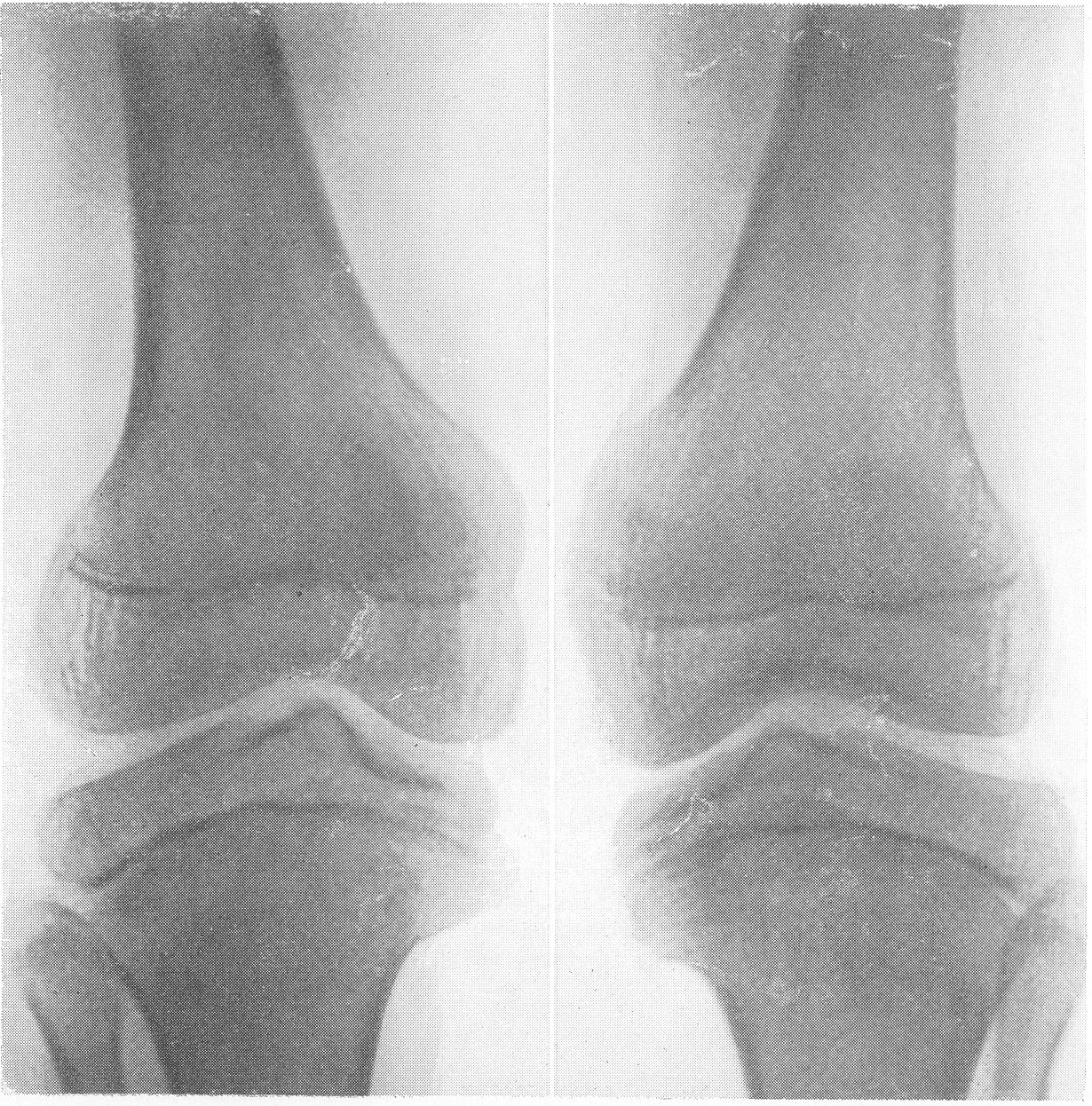
Этот вид карликового роста представляется чрезвычайно характерным, и хондродистрофика можно узнать с первого взгляда. Конечности по сравнению с нормальными размерами туловища и головы непропорционально коротки, т. е. имеется микромелия. Череп брахицефаличен, иногда гидроцефалического типа. Лицо Плоское, лоб крутой, спинка носа втянута, так что все хондродистрофики необычайно похожи друг на друга. У новорожденных определяется кифоз позвоночника; у детей, уже ходящих, и у взрослых имеется кифотическое искривление грудного и верхнепоясничного отдела позвоночного столба, а нижнепоясничный отдел позвоночника показывает характерный лордоз. Живот выпячивается вперед, и в профиле ягодицы резко выдаются cзади. Клинический диагноз хондродистрофии обычно настолько прост, что нет надобности в других способах распознавания. Рентгенологическое исследование необходимо с научно-исследовательской точки зрения и для детального изучения всех особенностей пораженного скелета. Трубчатые кости на рентгенограммах коротки и неуклюжи, они кажутся относительно утолщенными, а в некоторых случаях их поперечник и в действительности больше, чем у нормальных ровесников (рис. 262, Г; 269—272). Корковый компактный слой всегда утолщен, особенно в средней части диафиза.
{kind=link}
Кости очень плотны и дают очень интенсивную тень. В результате несоразмерной нагрузки большого торса на короткие конечности и тракции хорошо развитых крепких мышц кости обычно несколько дугообразно искривлены: это особенно касается бедренной кости в нижней ее трети, а также плечевой кости. Все нормально преформированные бугры, выпячивания и шероховатости на поверхности кости, главным образом места прикрепления мышц, значительно утрированы и грубы. Таковы, например, вертелы бедра, особенно малый, дельтовидная шероховатость плеча, бугристость большеберцовой кости, лучевой кости, локтевой отросток локтевой кости и т. д. Очень характерным признаком хондродистрофической деформации считается участие сравнительно мало пораженной малоберцовой кости в образовании коленного сустава. Малоберцовая кость имеет длину большеберцовой кости или превышает ее и не сочленяется, как в норме, с основанием наружного мыщелка большеберцовой кости, а выступает кверху (рис. 269). Нередко имеется genu varum. Головка плеча из-за больших размеров большого бугорка сидит сбоку кости, и плечо имеет вид humerus varus. Деформирована и недоразвита также головка бедра. Метафизарные концы костей имеют форму раструба, они вздуты наподобие бокала или канделябра, их компактные края заострены. Массивные, неуклюжие и расширенные в поперечнике эпифизы в виде грибов нависают над метафизами. Зона предварительного обызвествления метафизов обычно расширена и подчеркнута. Она дает не гомогенную линейную тень, а разбита на ряд более или менее близко отстоящих друг от друга продольных темных костных полосок.
{kind=link}
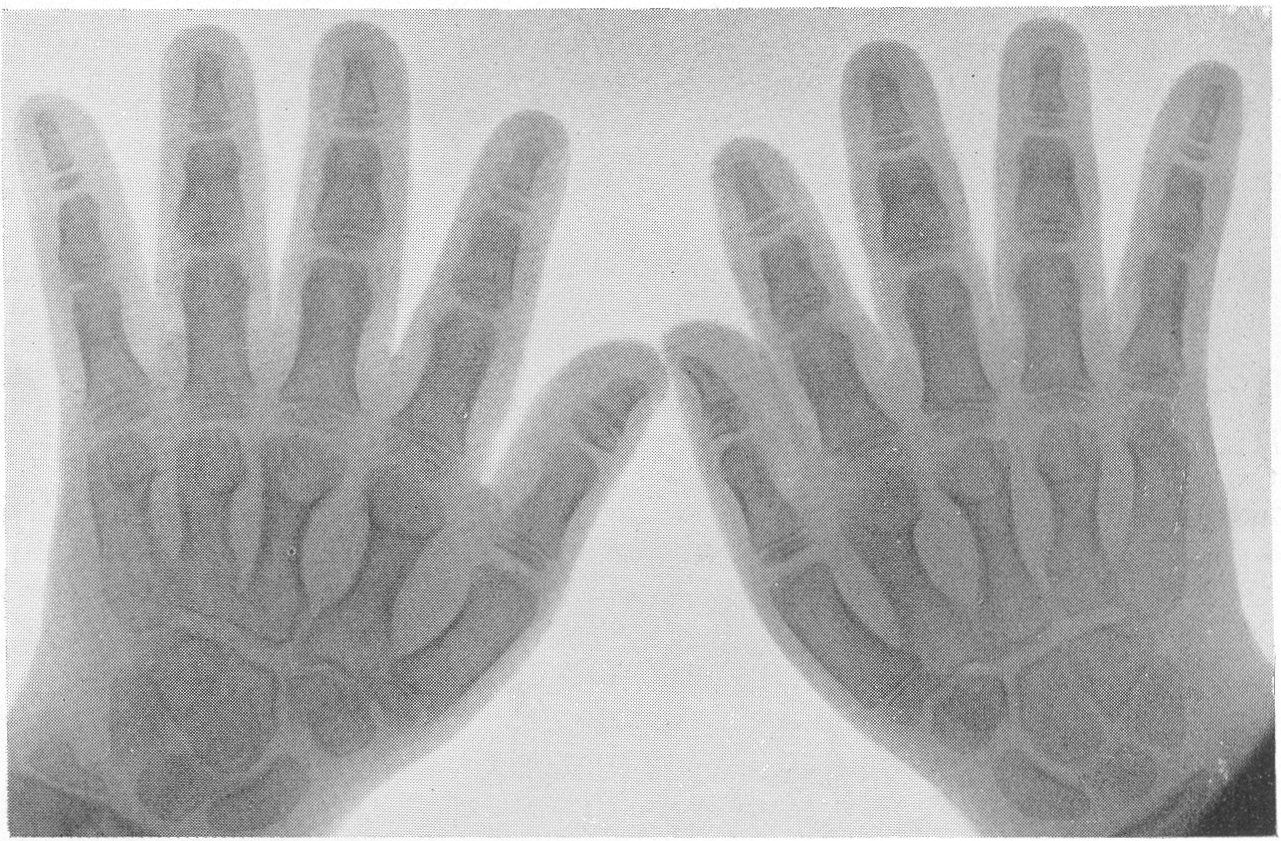
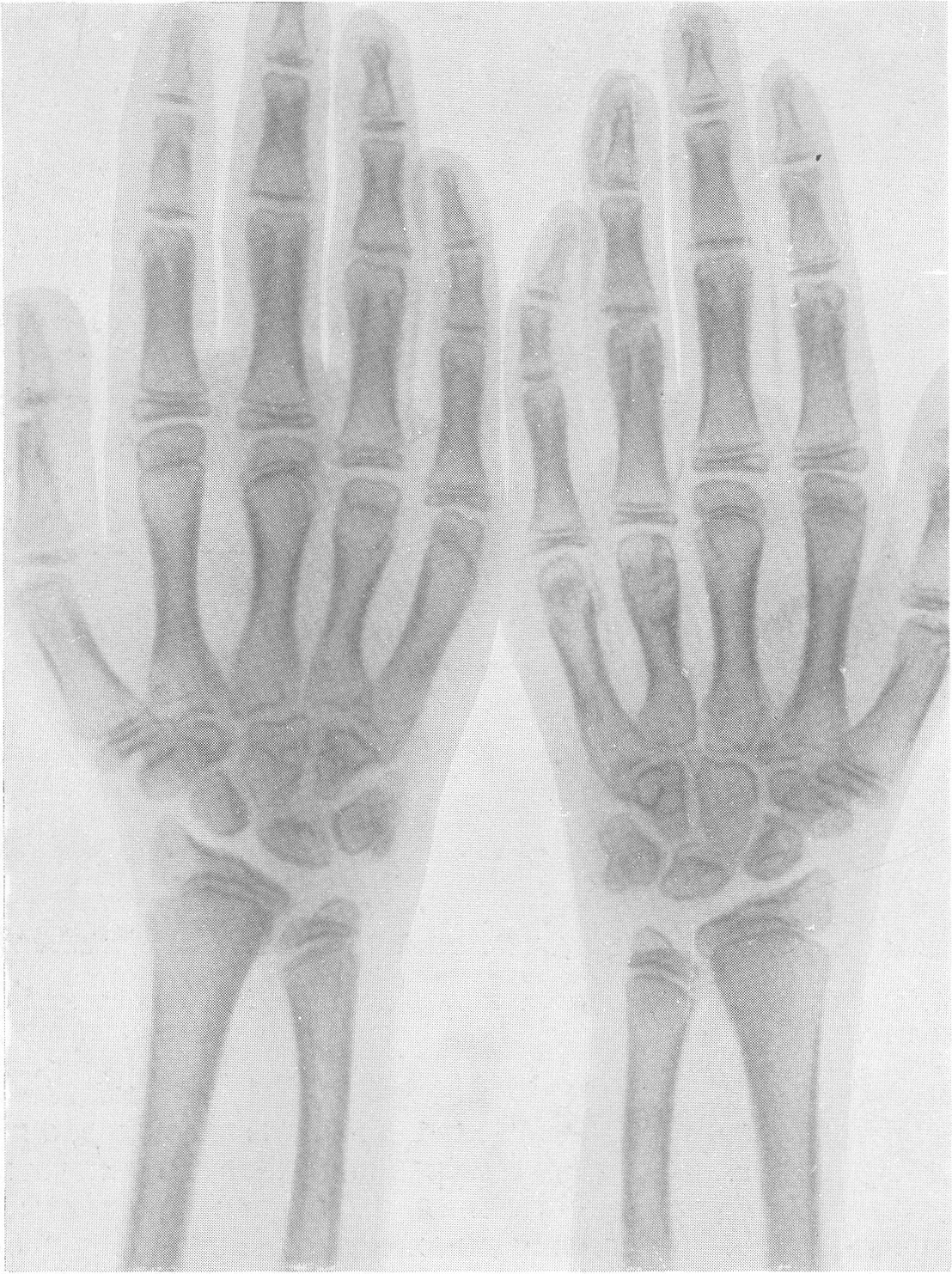
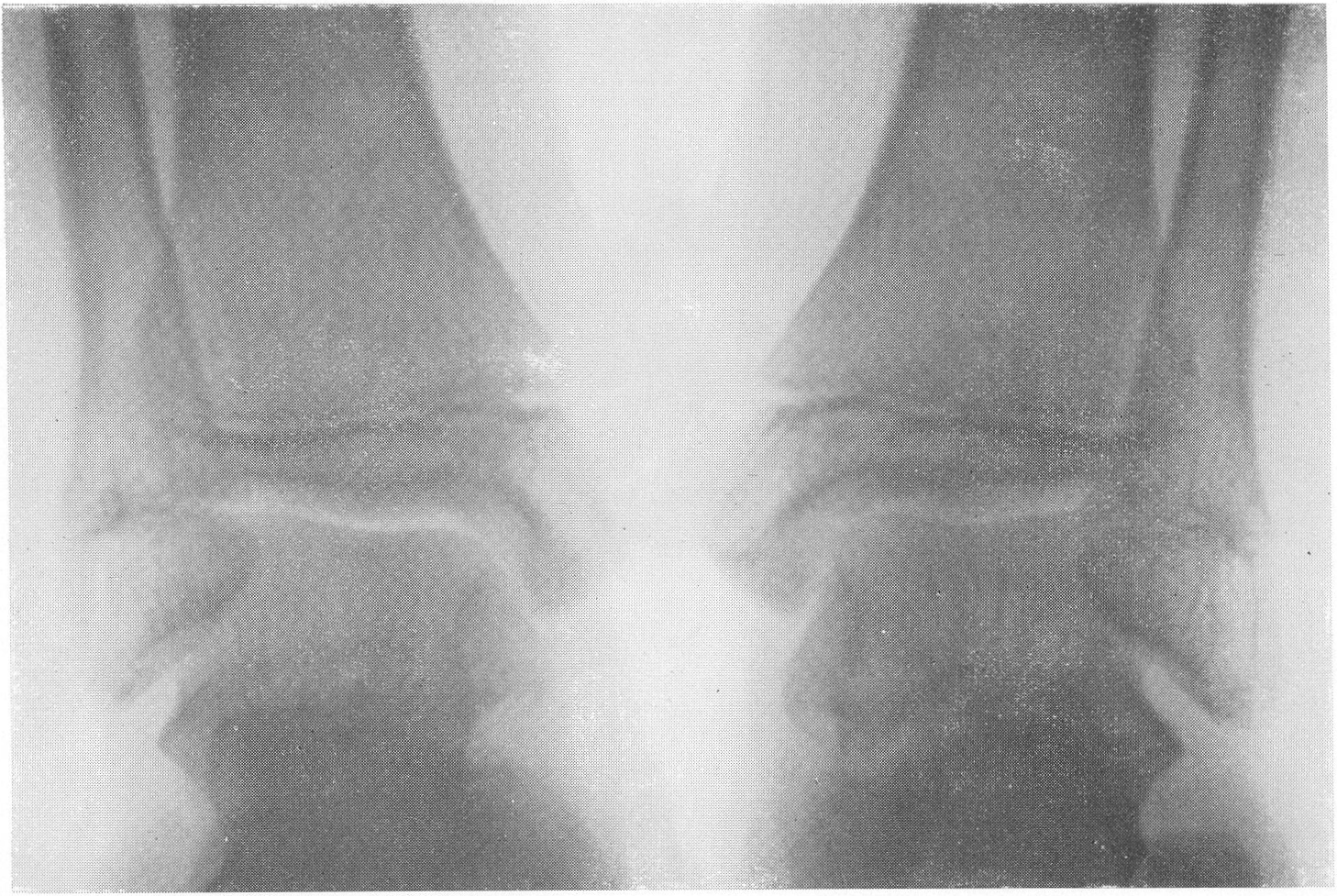
Эпифизарные ядра окостенения в общем появляются в срок, отдельные точки окостенения становятся заметными то несколько преждевременно, то запаздывают. Ядра окостенения имеют не гладкие резко ограниченные контуры, а деформированы, неровны, изгрызаны и бахромчаты. Свет лая эпифизарная линия проходит не строго перпендикулярно к длиннику костей, а наискось, в большинстве случаев она дугообразно искривлена по направлению к метафизу. Ширина эпифизарной хрящевой зоны очень колеблется, так что эпифизарное ядро лежит то совершенно отдельно в блюдцеобразном вдавлении метафиза, то сливается с тенью метафиза и едва выступает на рентгенограммах. Структурная сеть губчатого костного вещества эпифиза значительно рыхлее и шире, чем в метафизе. Поэтому эпифизы выделяются своей менее интенсивной тенью, и все эпифизарные концы костей на рентгенограммах более светлы, чем диафизы. Закрытие ростковых зон происходит обычно раньше срока, т. е. в конечном счете имеется ускорение энхондрального окостенения. Крайне типичную, патогномоничную картину представляет кисть хондродистрофика (рис. 270 и 271). Все пальчики укорочены и кажутся обрубленными. Кроме того, они имеют одинаковую длину. II, III, IV, V пальцы, в особенности первые три, лежат не параллельно друг другу, а расходятся лучеобразно, и их обычно сравнивают с трезубцем.
{kind=link}
Костные фаланги имеют почти квадратную форму, они в 2 или 3 раза короче, чем при нормальных условиях. Основание черепа вследствие преждевременно произошедшего костного соединения в synchondrosis intersphenoidalis и sphenooccipitalis на боковом снимке представляется укороченным. Блуменбахов скат стоит круто. Гипофизарная ямка имеет обыкновенно малые размеры и уплощенную форму. Processus clinoidei плохо обрисовываются или вовсе не развиты. Плоские кости свода черепа вследствие укорочения основания относительно увеличены и как бы нависают над лицевым черепом. Придаточные полости носа хорошо пневматизированы. Позвоночник не представляет резких рентгенологических изменений. Тела могут быть несколько уплощены, в особенности тела крестцовых позвонков, иногда наблюдается генерализованная бревиспондилия. Суставные поверхности межпозвонковых суставов очень неровны. Все размеры таза уменьшены, таз сужен, края подвздошных гребней также неровны, светлые крестцово-подвздошные сочленения очень широки, запирательные отверстия увеличены. Вертлужная впадина уплощена и поверхность ее извилиста. Ребра по сравнению с другими длинными костями мало, но все же укорочены, они располагаются горизонтально, и грудная клетка несколько уплощена и широка. Грудинные концы ребер, а также головки и шейки утолщены и деформированы. Ключицы идут совершенно прямо и не показывают нормальных искривлений. Из всех костей скелета именно ключицы менее всего изменены при хондродистрофии. Это исключительное положение вытекает из сравнительно раннего окостенения ключиц и их соединительнотканного происхождения. Рентгенологические признаки хондродистрофии настолько типичны, что дифференциальной рентгенодиагностики в сущности практически и нет. Трудности возникли, можно сказать, искусственно, когда понятие о хондродистрофии некоторыми авторами стало истолковываться расширительно и был поднят вопрос о так называемых атипичных формах хондродистрофии. К этим атипичным формам хондродистрофии некоторые клиницисты, да и рентгенологи без достаточного основания стали причислять всевозможные конституциональные варианты и врожденные пороки развития и уродства, сопровождающиеся относительно короткими конечностями, т. е. чуть ли не любую микромелию. Сейчас окончательно установлено, что основную массу так называемых атипических форм хондродистрофии составляют больные, страдающие совершенно самостоятельной нозологической формой, а именно остеохондродистрофией, с которой и требуется произвести дифференциальную диагностику, в частности и в особенности именно рентгенодиагностику. Исключить надо также несовершенный остеогенез — у новорожденных и маленьких детей, так как у детей старшего и уж подавно у взрослых смещение все более расходящихся по различным путям развития болезней не может происходить. В виде исключения отличительного распознавания требуют некоторые проявления рахита и кретинизма, а также врожденная эпифизарная точечная костная дисплазия. Несколько особняком стоит уровская болезнь (болезнь Кашина—Бека). Но все это просто с рентгенологических позиций, при условии элементарного знакомства с каждой из перечисленных болезней в частности.
3. ХОНДРОЭКТОДЕРМАЛЬНАЯ ДИСПЛАЗИЯ (СИНДРОМ ЭЛЛИС—ВАНКРЕВЕЛЬДА)
Хондроэктодермальная дисплазия, или синдром Эллис—Ванкревельда (Ellis, S. van Creveld), — это очень редкая врожденная дисплазия, сказывающаяся уже у новорожденных и в раннем детском возрасте, при которой, кроме изменений костной системы, налицо еще неправильности других систем эктодермального происхождения. Этот синдром составляет тетрада, а именно, во-первых, „хондродисплазия”, т. е. нарушение костеобразования из хрящевой основы, что выражается в абсолютном и относительном укорочении конечностей, главным образом дистальнее колен и локтей. Поэтому характерна низкорослость. По какой-то непонятной причине особенно укорачивается малоберцовая кость. Имеется также недоразвитие вертлужной впадины. Во-вторых, при синдроме Эллис—Ванкревельда обязательна полидактилия, а иногда и синдактилия и синметакарпия. Обычно имеется по 6 пальцев, причем лишним является палец с ульнарной стороны. В-третьих, налицо недоразвитие, истончение ногтей, зубов, скудные редкие волосы. В-четвертых, в этот синдром входит врожденный порок сердца, чаще всего дефект межжелудочковой перегородки. Рентгенологически можно уточнить, что имеется замедленный и извращенный процесс энхондрального окостенения, трубчатые кости коротки и толсты, могут выявиться и слияния костей запястья (головчатой и крючковатой), пропорции костей нарушены.
4. ОСТЕОХОНДРОДИСТРОФИЯ
Эта недостаточно известная в широких врачебных кругах врожденная системная болезнь скелета встречается, по нашим данным, отнюдь не так уж редко, во всяком случае чаще популярной, во всех учебниках представляемой и поэтому и хорошо распознаваемой хондродистрофии. Остео хондродистрофия — это, несомненно, вполне самостоятельная, весьма четко очерченная и характерная нозологическая форма. Она стала предметом углубленного научного изучения лишь в поздних 20-х годах текущего столетия благодаря рентгенологии. На страницах нашей отечественной печати первое квалифицированное описание этой болезни дали в 1928 г. Е. С. Рабинович и И. А. Мухин. В 1936 г. этой болезни как атипической форме хондродистрофии посвятили журнальную статью Д. Г. Рохлин и Е. П. Велицкий. В 1939 г. на эту тему защитил кандидатскую диссертацию А. А. Смирнов. Е. С. Рабинович и И. А. Мухин изучили эту болезнь почти одновременна с Сильвершельдом (Silverskiold), Брейлсфордом (Brailsford), Грудзинским (Grudzinski), Моркио (Morquio) и другими иностранными авторами, именами которых обозначается эта болезнь. В старой литературе можно найти немало несомненно сюда относящихся разрозненных казуистических сообщений, однако неправильно толкуемых и недостаточно полно изученных, особенно с точки зрения современных строгих рентгенологических требований. Как это всегда бывает со сравнительно новыми нозологическими формами, их изучение проходит через обязательную фазу первоначального накопления формально-описательного казуистического материала, и в литературе неминуемо накапливается множество самых разнообразных названий. Их надо на первых порах знать, чтобы в этой мешанине правильно разобраться — объединить одинаковое, однозначное и разъединить различное, случайное, не относящееся к существу. Мы предлагаем пользоваться пусть весьма условным, но все же наиболее простым названием „остеохондродистрофия”. Е.С. Рабинович и И. А. Мухин говорили лишь глухо о „своеобразном системном поражении костей”, т. е. по существу сознательно отказались от определения понятия. Неудачными мы считаем применяемые в литературе и даже теми или иными авторами настойчиво пропагандируемые названия, как генерализованные остеохондропатии с симметричной гипоплазией суставов, симметричные гипоплазии суставов, системные остеохондропатии (в кавычках или без кавычек), множественная эпифизарная дисплазия, энхондральный мета-эпифизарный дизостоз, спондило-эпифизарная хондродисплазия, множественные местные маляции, деформирующая наследственная (или семейная) хондроостеодистрофия, платибрахиспондилия, множественная системная платиспондилия, атипическая хондродистрофия, forme fruste (т. е. невыраженная, неполноценная форма) хондродистрофии и т. д. Несправедливо и исторически неправильно поддерживать такие названия, как болезнь Моркио, болезнь Моркио—Брейлсфорда и т.д. Остеохондродистрофия наблюдается спорадически и неожиданно, то в совершенно здоровых семьях, то нередко имеет явно семейный характер, т. е. этой болезнью поражаются подчас несколько или даже много членов одной и той же семьи, притом в различных сочетаниях, например в одном, двух или больше поколениях. Рентгенологу приходится видеть больных, страдающих остеохондродистрофией, как и хондродистрофией, в любом возрасте. Но основной контингент наших больных все же относится к детскому возрасту, когда у родителей заболевших еще имеется надежда на врачебную помощь и возможность успешного лечения, а у врачей с наибольшей остротой возникают диагностические и дифференциально-диагностические сомнения и трудности, побуждающие искать консультации рентгенолога. Клиническую картину остеохондродистрофии мы считаем очень яркой. Прежде всего бросается в глаза малый рост, обусловленный укорочением туловища. Позвоночный столб, а за ним и весь торс сокращается по сравнению с нормой примерно на треть своей высоты, а в тяжелых случаях — и до половины! Конечности же почти совсем нормальны по длине или же, в случае их поражения, кажутся непропорционально длинными из-за короткого торса. Сказать, что остеохондродистрофики — карлики, это преувеличение. Области суставов заметно утолщены. Укорочена и шея. Поэтому нормальная по размерам голова посажена очень низко, она как бы сидит на плечах, а в тяжелых случаях подбородок покоится на груди, на грудине. Особенно характерный вид являют больные остеохондродисторофией при осмотре сбоку. Больной согнут „в три погибели ”, укороченный торс резко увеличен в переднезаднем разрезе, выстоят вперед грудина и нижнепередние края ребер, у ребят торчит вперед большой животик, кзади выдаются спинка и ягодицы. Кифоз типичен: он располагается на месте перехода грудного отдела позвоночника в поясничный, сильно выражен, так что в резко выраженных случаях изгиб кзади может достигнуть степени углового горба почти под прямым углом. Конечности, особенно нижние, деформированы. Ноги несколько согнуты в тазобедренных и коленных суставах, как правило, строго симметрично. Колени обычно вальгированы. Имеется плоскостопие. В связи с этим характерна неуклюжая покачивающаяся походка — утиная, вразвалку. Изменены и руки, вернее, пальцы: они равномерно видимо коротки, с широкими свободными концами, как бы обрублены на одном уровне — так называемая изодактилия. Изменения суставов различны: в ряде случаев наблюдается ограниченная подвижность суставов, особенно крупных. Нередко же, наоборот, из-за слабости сумок и связочного аппарата суставы разболтаны, что чаще всего происходит с периферическими малыми суставами, т. е. пальцев рук и ног, подчас и с лучезапястными и голеностопными. Мускулатура развита слабо. Остеохондродистрофия, подобно несовершенному остеогенезу, в каждом индивидуальном случае выражена в различной степени по объему и интенсивности поражений скелета. Иногда болезнь так сильна, что наступает внутриутробная смерть. Но в подавляющем большинстве практически наблюдаемых нами случаев ребенок в первые годы жизни может ничем не отличаться от своих нормальных сверстников, и врожденная болезнь до поры до времени внешне себя ничем еще не проявляет. Более тяжелые случаи становятся у выживающих детей явными, когда ребенок становится на ножки и начинает ходить, и первым признаком заболевания тогда служит развивающийся на типичном месте безболезненный кифоз или горбик. Но чаще всего, по нашим наблюдениям, проходят нормально первые 5—6 лет врожденной болезни, а затем уже начинает меняться внешний облик ребенка. Он постепенно становится сутулым, коротышкой. Ребенок начинает жаловаться на небольшие боли в спинке, а также в области крупных суставов — тазобедренных и коленних. Прогрессирует общая слабость, дети или подростки подолгу лежат малоподвижными, вялыми, нарастает мышечная слабость, адинамия. Походка становится все более скованной и затруднительной, так что в тяжелых случаях больные в состоянии передвигаться только с помощью посторонних. В подобных случаях какая-нибудь привходящая детская болезнь, с которой нормальный ровесник справляется, способна стать причиной смерти у больного остеохондродистрофией. Но вероятно, что болезнь прекращается или как-то самопроизвольно с наступлением половой зрелости затихает, и немалое число больных доживает до зрелого возраста. Вместе с тем рост и развитие больного может приостановиться в любой фазе детства, и тогда больной может превратиться в подлинного карлика, что, впрочем, наблюдается сравнительно редко, и лучше говорить при остеохондродистрофии о низком или малом росте, а не о карликовости.
{kind=link}
Рис. 273. Остеохондродистрофия у 13-летнего мальчика с типичной клинико-рентгенологической картиной заболевания. Рентгенограмма плечевой кости.
Лицо больных с остеохондродистрофией ничем не выделяется, оно обычное, выражая нормальное или изредка лишь несколько задержанное умственное развитие. Оба пола поражаются с одинаковой частотой. Со стороны различных систем определенных нарушений при неосложенной остеохондродистрофии не бывает. В редких случаях указывают на толстую неэластическую кожу, и дело может доходить до склеродермии, затрудняющей движения пальцев — при разболтанных суставах! Иногда волосы бывают сухими, жесткими. Упоминаются также весьма редкие нарушения со стороны глаз — помутнения роговицы, атрофия зрительных нервов, прогрессирующая потеря зрения. Эндокринная система явных уклонений от нормы не представляет. Нервная система никем до сих пор не подвергнута специальному изучению. Этиология остеохондродистрофии и ее патогенез — загадка, и мы не будем здесь перечислять многочисленные предположения, которые высказаны на этот счет. Сущность болезни — это какое-то глубокое врожденное нарушение процессов превращения хряша в костную ткань, т. е. своеобразная неполноценность энхондрального, но не периостального окостенения. Под микроскопом дегенеративные изменения в хрящевой энхондральной системе не представляют ничего специфического. В полном соответствии со всеми этими данными находится богатая рентгенологическая картина остеохондродистрофии. Все кости скелета в небольшой степени поротичны. Диафизы больших трубчатых костей (рис. 261, Д) грубых изменений не представляют. Они чуть толще обычного. Поэтому они кажутся короче нормальных. Впрочем, подчас они у наших больных и действительно укорочены, особенно бедренная и плечевая кости по сравнению с голенью и предплечьем. При этом корковый слой диафизов немного истончен, костномозговой канал широк, и только по линиям повышенной нагрузки корковый слой плотен и толст (рис. 273). Суставные концы костей расширены. Эпифизы неправильной конфигурации, неуклюжи, увеличены в поперечнике, но уплощены, угловаты.
{kind=link}
Рентгенограмма области обоих коленных суставов. Малоберцовая кость стоит на нормальной высоте. Надо полагать, что при остеохондродистрофии укорочение костей по существу и приосходит за счет снижения высоты эпифизов, а не укорочения метадиафизов. Наружные контуры эпифизов шероховаты, а то и изъедены. Отдельные ядра окостенения расщеплены, разбиты на части, фрагментированы, т. е. появляется множество неправильных бесформенных островков окостенения. Когда эпифизы с возрастом развиваются, „созревают”, структура их еще в какой-то степени упорядочивается, но деформация остается стойкой и необратимой. Широки и подчеркнуты в виде плотных лент зоны предварительного окостенения (рис. 274). Так же изменены и малые трубчатые кости. Фаланги немного укорочены и утолщены, их губчатое вещество имеет своеобразную мелкоячеистую структуру (рис. 275 и 276). Темпы окостенения в нашей серии случаев несколько замедлены, ядра окостенения в обычных случаях появляются с небольшим опозданием. Могут развиваться псевдоэпифизы. Поэтому у маленьких детей интервалы между метафизами противостоящих костей велики — рентгеновские суставные щели порой чуть ли не в 2 раза шире нормы. Окончательное слияние эпифизов с диафизами происходит с уклонениями от нормальных сроков в ту и иную сторону, иногда с большими колебаниями. Особенно большие перемены наблюдаются у детей в школьном возрасте, то раньше, то позже, в костях тазобедренного сустава (рис. 277 и 278). Эпифизарная головка бедра сперва может долго заставлять себя ждать. Появившись, она в определенном юношеском возрасте приобретает неправильную форму и структуру. Она увеличивается в размерах, уплощается и расширяется, распадается на множество неправильных ландкартообразных глыбок.
{kind=link}
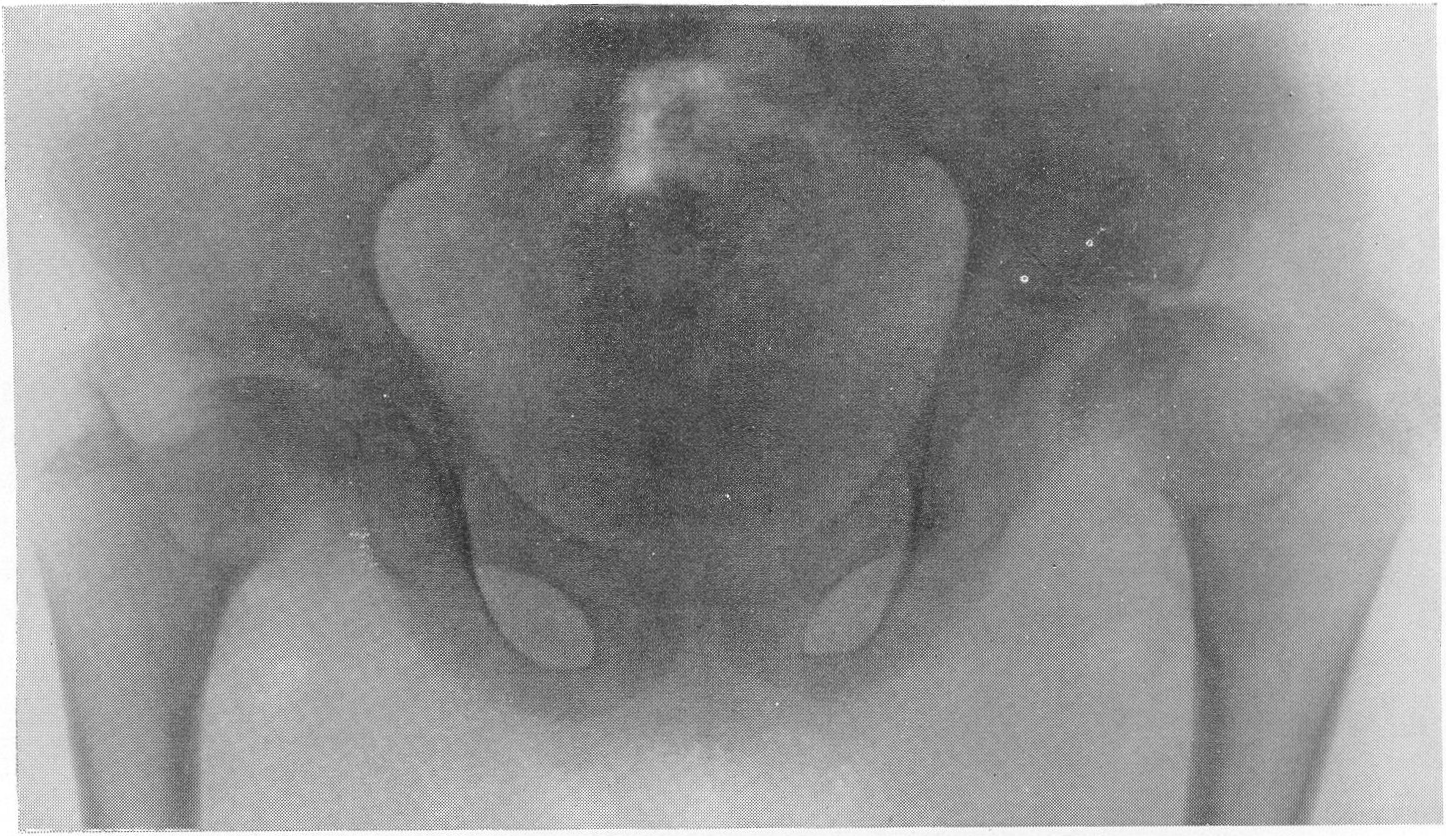
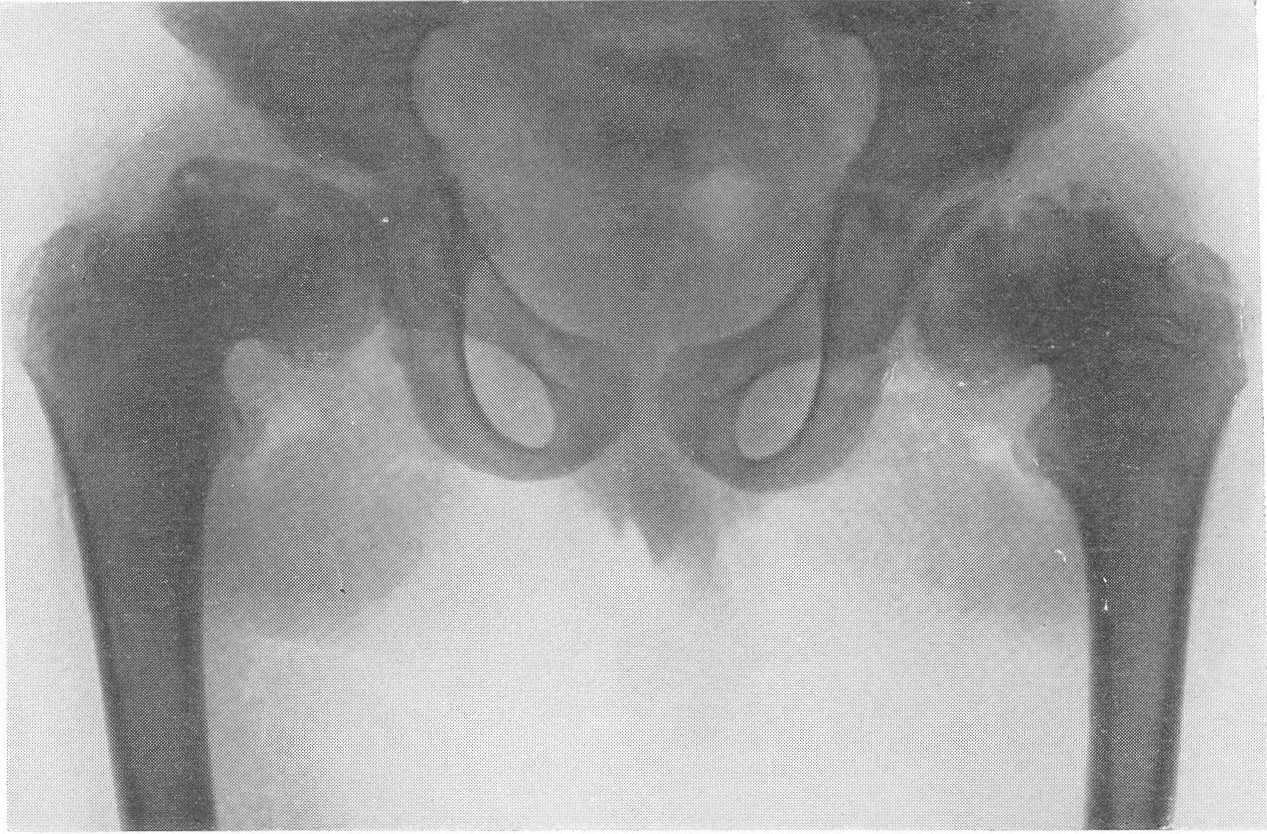
Шейка бедра сильно укорачивается и утолщается, большой вертел смещается высоко, другими словами, развивается варусная деформация. Вертлужная впадина также приобретает неправильную форму, расширяется, уплощается и обезображивается, суставные элементы тазобедренного сустава теряют свое взаимное соответствие. Все это придает изменениям при остеохондродистрофии очень большое сходство с остеохондропатией бедренной головки. Таз в целом также претерпевает уклонения от нормы, он запрокидывается, сдавливается с боков и в особенно тяжелых случаях остеохондродистрофии тазовое кольцо приближается к форме пресловутого карточного сердца или винного бокала. Для области коленного сустава отличительным признаком является, как уже сказано выше, вальгусная деформация.
{kind=link}
{kind=link}
Рис. 277. Та же больная. Рентгенограмма таза. Типичные изменения бедренных головок, напоминающие остеохондропатию.
Значительная варусная деформация. Ребенок направлен на рентгенологическое исследование в связи с хромотой в последний год: был заподозрен односторонний туберкулезный коксит.
{kind=link}
Рис. 278. Та же больная. Рентгенограмма области таза. Картина, внешне напоминающая двустороннюю остеохондропатию.
Она выражается в разнице в высоте эпифизов бедра, а именно высота медиальной части эпифиза бедра заметно выше латерального, и колени расходятся в виде буквы „X”. Головка малоберцовой кости стоит на нормальном уровне и, следовательно, в образовании коленного сустава участия не принимает.
{kind=link}
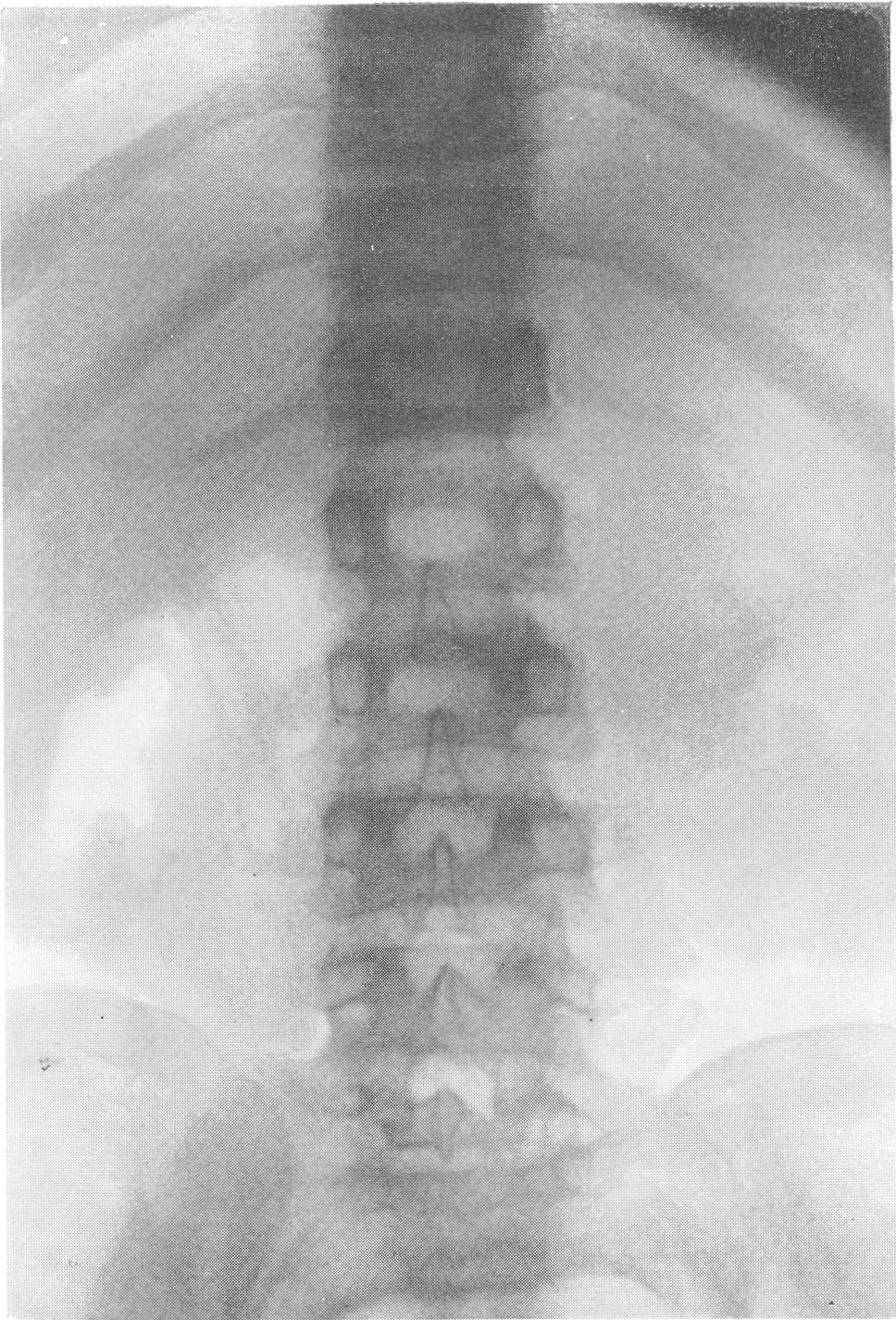
На рентгенограммах скелета грудной клетки обращает на себя внимание ее призматическая форма, грудина своим нижним концом выпячена вперед, ключицы приподняты. Ребра утолщены, очень широки и плоски. Они сильно придвинуты друг к другу, т. е. межреберья сужены. Диафрагма и поддиафрагмальные органы смещены вверх, в краниальном направлении. Что же касается позвоночного столба, то здесь патологическая рентгенологическая картина особенно выражена и постоянна (рис. 279 и 280). Все без исключения тела позвонков имеют уменьшенную высоту, уплощены, т. е. налицо системная брахиспондилия. К сожалению, часто говорят и пишут о платиспондилии, что означает не только снижение высоты тела, но и подлинное, а не кажущееся увеличение поперечных размеров тела позвонка. Истинное и притом значительное укорочение всего позвоночного столба происходит за счет костной части его, — межпозвонковые хрящевые диски особых изменении не представляют. Сочленовные отростки, как и поперечные и остистые отростки, коротки и неуклюжи.
{kind=link}
{kind=link}
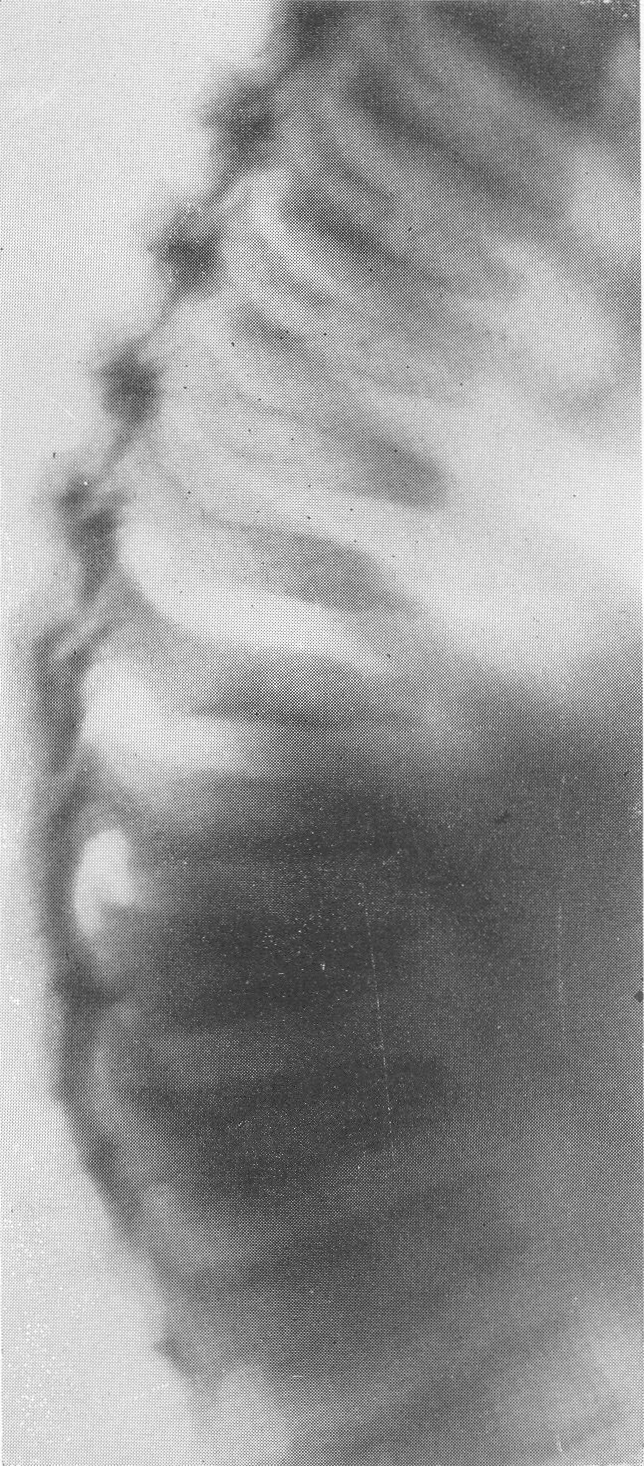
Деформация тела одного из верхних поясничных позвонков в боковой проекции, крайне типичная для остеохондродистрофии и гаргойлизма — ангуляция позвоночника. Тип деформации по Пфаундлеру—Гурлер („нижний язык”). Основное заболевание осложнено успешно леченным рахитом; полосы склероза. Структура позвонков поротична, грубопетлиста. Контуры тел позвонков подчеркнуты, но теряют гладкий ход, становятся неровными. Самым характерным рентгенологическим проявлением остеохондродистрофии служит определенная, ни с чем не смешиваемая и диагностически важная деформация, обрисовывающаяся на боковых снимках (рис. 281—283). Речь идет о том, что один или два позвонка, а именно на месте перехода грудного отдела в поясничный — чаще всего I или II поясничный позвонок, иногда и XII грудной позвонок, спереди показывают изъян. Верхняя половина позвонка недоразвивается, нижняя половина выдается вперед, как нос корабля, и поэтому спереди тело ступенчато или клювовидно деформировано, оно принимает клиновидную, заостренную кпереди форму, верхнепередний контур становится лестничным, террасоподобным.
{kind=link}
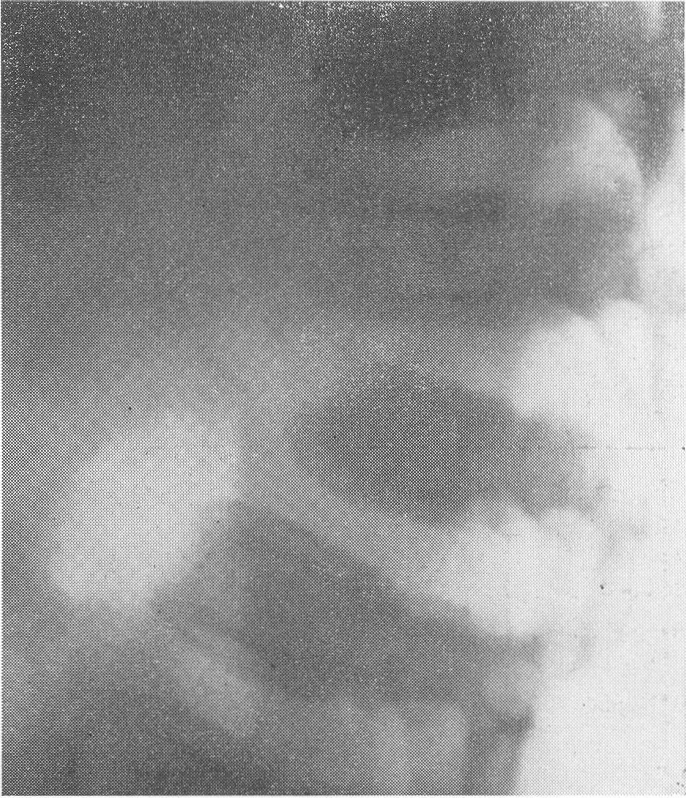
Горб с выстояннем остистых отростков в пояснично-грудном отделе. Типичная деформация тела XII грудного позвонка. Тип деформации по Моркио—Брейлсфорду („центральный язык”). Ранний вторичный значительный деформирующий спондилоз. Это так называемый первый тип деформации — тип Пфаундлера—Гурлер (М. Pfaundler, Gertrude Hurler), „нижний язык”, чаще всего наблюдаемый при гаргойлизме (см. ниже) (рис. 281, 283, А). В других же случаях укороченное тело позвонка вытягивается и заостряется посередине, на равном расстоянии от верхней и нижней площадок, это так называемый второй тип деформации — Моркио—Брейлсфлода, «центральный язык» (рис. 282, 283, Б). В некоторых случаях эта особенно качественная, характерная для остеохондродистрофии деформация происходит в телах не одного, а нескольких соседних позвонков. Этот уменьшенный в размерах недоразвитый позвонок тогда как бы выскальзывает из правильного ряда кзади, а вышележащие грудные позвонки над ним нависают. Так развивается специфическая для остеохондродистрофии деформация позвоночного столба — кифоз или ангуляция, т. е. угловой изгиб, горбик. При очень резком угловом искривлении может происходить сдавление задней поверхностью тела смещенного позвонка спинного мозга, сопровождающееся соответствующими тяжелыми вторичными, опасными для жизни неврологическими последствиями. В черепе рентгенологически ничего особенного не определяется. Он кажется лишь относительно большим. Область турецкого седла нормальна. В тяжелых случаях может быть некоторая степень конвексобазии, позвоночник вдавливает основание черепа кверху, что еще более усугубляет укорочение шеи и приближение черепа к грудной клетке. Следует также обратить внимание на сравнительно частый сильно развитый кариес зубов.
{kind=link}
Рис. 283. Схема типичной для остехондродистрофгш деформации XII грудного, I или II поясничного позвонка, чаще наблюдаемый при гаргойлизме. Тип А по Пфаундлеру — Гурлер, „нижний язык”; тип Б по Моркио— Брейлсфорду, „центральный язык”. При первом типе имеется пологий скат верхней площадки, при втором типе — два ската, верхней и нижней площадок тела пораженного позвонка.
Остеохондродистрофия имеет высокий дифференциально-диагностический потенциал. Это понятно: она проявляется в каждом индивидуальном случае в весьма широких и разнообразных по глубине и по охвату пределах, она поражает в разнообразных сочетаниях различные отделы скелета и к тому же приобретает практический интерес в самых различных возрастных условиях, выражая при этом свойственные каждой отдельной возрастной группе клинико-рентгенологические особенности. Этот полиморфизм не может не породить обстановку для смешения остеохондродистрофии с большим рядом общих и местных костных патологических процессов. Надо также всегда иметь в виду, что остеохондродистрофия может протекать вообще несколько лет жизни без достаточно ясных внешних клинических проявлений, и ее врожденный характер может поэтому оставаться довольно долго нерасшифрованным и недооцененным. А между тем рентгенологическое исследование способно в любой момент этого клинически бессимптомного или кажущегося бессимптомного периода благодаря богатой симптоматологии обеспечить точное раннее распознавание. Прежде всего — об отличительном распознавании с хондродистрофией. У новорожденного, страдающего хондродистрофией, вся основная патология уже налицо, а остеохондродистрофия может в этом периоде жизни еще ничем внешне не выражаться. В дальнейшем, при первом взгляде на болеющего остеохондродистрофией (ребенка или юношу, а тем более взрослого больного), да и на его рентгенограммы, есть что-то общее с хондродистрофией. Но при более внимательном изучении симптомов этих обеих врожденных и симметрично развивающихся болезней становятся ясными и гораздо более резкие отличительные черты. Общим является нарушение правильных пропорций тела, разграничивающим же — совершенно различный характер диспропорции, а именно длинное туловище и короткие конечности при хондродистрофии и, наоборот, короткое туловище и длинные конечности при остеохондродистрофии. При хондродистрофии никогда не бывает системной бреви- или платиспондилии, обязательной и постоянной для остеохондродистрофии, уже не говоря об ангуляции пояснично-грудного отдела позвоночника, столь характерной для остеохондродистрофии. Некоторые весьма тонкие дифференциально-диагностические рентгенологические признаки в картине таза и пояснично-крестцового отдела позвоночника разработал в 1958 г. Кеффи. Трезубец резко укороченных пальцев кисти хондродистрофика так же клинически и особенно рентгенологически при элементарном знакомстве с обеими болезнями легко отличим от изодактилии неукороченных пальцев при остеохондродистрофии. Различны при обоих заболеваниях рентгенологические детали формы и структуры черепа, суставов вообще и тазобедренных и коленных суставов в частности. Поэтому с современных научных позиций надо раз и навсегда оставить путаные представления о переходных формах, об атипичных случаях хондродистрофии и не следует включать в старый строго очерченный круг хондродистрофии другую самостоятельную болезнь, каковой является остеохондродистрофия. Эти обе болезни могут и должны быть строго различимы. Дифференциальной диагностики требуют и такие общие заболевания скелета, как приобретенная уровская болезнь (Кашина—Бека) и кретинизм (общий гипотиреоз). Особенно важно не принять за остеохондродистрофию кретинизм и вообще эндокринные нарушения, так как при эндокринных расстройствах диагностическая ошибка чревата серьезной ошибкой терапевтической. Ведь если мы в настоящее время не в состоянии влиять на течение остеохондродистрофии, то при гипотиреозе и других эндокринопатиях специальная гормональная терапия способна давать весьма положительные лечебные результаты. Подробнее о дифференциальной диагностике целесообразно говорить после детального изучения этих представителей костной патологии (кн. 2, стр. 60). Один из наших случаев остеохондродистрофии был направлен к нам на консультативное заключение с предполагаемым диагнозом болезни Реклингхаузена, что является грубой ошибкой. Так же нет в сущности никаких серьезных оснований для смешения остеохондродистрофии с остеопойкилией (стр. 451). Далеко зашедшие случаи остеохондродистрофии, ведущие подчас к очень низкому росту, следует включить в общий сложный и обширный круг болезней, требующих детального разбора причин отставания роста и карликовости. Об этом — также подробнее в дальнейшем, когда станет яснее большое многообразие форм карликового роста. Из общих заболеваний легко исключить системные поражения костей при нарушениях ретикуло-эндотелиального и кровотворного аппарата, выделительных органов и т. д., так как у больных остеохондродистрофией морфологически и биохимически кровь нормальна, а внутренние органы (печень, селезенка, почки, кишечник и пр.), костный мозг и лимфатическая система патологических изменений не представляют. Увеличенные в объеме узловатые области всех суставов могут направить клиническую мысль по ложному пути предположений о полиартрите. И здесь рентгенологическое исследование способствует уточнению диагноза. При знакомстве с остеохондродистрофией тогда не возникнет соблазна говорить и писать о всевозможных „новых” болезнях под выдуманными названиями, как «минимальные», «частичные» гипоплазии и дисплазии суставов, врожденные бестемпературные полиартриты, атипичные хондродистрофии с артритами, артрозами и т. п. Уж очень много казуистических сообщений развелось в литературе по этому вопросу, причем, как правило, эти „открытия” оказываются, попросту говоря, нераспознанными остеохондродистрофиями. Чаще всего больные, страдающие остеохондродистрофией, попадают на рентгенологическое исследование в связи с подозрением на ту или иную патологию скелета местного характера, т. е. когда на первый план в клинической картине выступают ограниченные деформации. Вот здесь главным образом и сказывается в первую очередь возрастной фактор. Всех наших больных остеохондродистрофией в раннем детском возрасте привело к рентгенологическому исследованию подозрение на туберкулезный спондилит или аномалию развития позвоночника. Клинициста смущает постепенно появившийся и медленно прогрессирующий кифоз или угловой горбик, протекающий сравнительно доброкачественно или совсем безболезненно, или же с умеренными болевыми нарушениями. Боковая рентгенограмма имеет решающее дифференциально-диагностическое значение, она показывает типичную картину на типичном месте, но для твердого и авторитетного исключения спондилита и местной врожденной неправильности требуется хорошее знакомство с остеохондродистрофией. В старшем детском, а именно в школьном возрасте, преимущественно в 10—12— 14 лет, больные направляются на рентгенологический контроль по другим диагностическим каналам, а именно как страдающие coxa vara, genua valga, но прежде всего остеохондропатией головки бедра, с которой — этого никто не отрицает — остеохондродистрофия получает величайшее внешнее рентгенологическое сходство. Особенно велика возможность ошибки при симметричном двустороннем остеохондропатическом процессе. К сожалению, немало ошибок вносит и рентгенологическое исследование — неизменно в результате одной и той же грубейшей и совершенно недопустимой методической ошибки, хорошо известной, но тем не менее слишком часто в жизни повторяемой. Мы имеем в виду рентгенологическое исследование одной только ограниченной области скелета, которая в данное время интересует клинициста. Непреклонный закон костной рентгенологии требует рентгенологического исследования, распространенного на весь скелет или во всяком случае на наиболее для каждого отдельного заболевания показательные его отделы, если только снимок ограниченной области хотя бы в ничтожной степени неясен, сомнителен, неубедителен. Целостное исследование скелета — вот простое и всем доступное средство предотвращения столь досадных и опасных ошибок при этой обширной дифференциальной рентгенодиагностике остеохондродистрофии. Практически, значит, никоим образом нельзя ограничиться исследованием одного лишь тазобедренного сустава, одного только таза в целом, одного колена, одной области или одной проекции позвоночника и т.д. Именно так многие „местные” заболевания и деформации сразу же оказываются тем, что они есть, т. е. лишь местным более заметным выражением общего системного заболевания. Таким образом, в спорном случае решает совокупность характерных черт остеохондродистрофии — диспропорциональный рост из-за уплощения позвонков и укорочения позвоночного столба с кифозом или угловым горбом на типичном месте, изменения суставных концов костей и прежде всего симметричные поражения элементов тазобедренных суставов у ребенка с нормальным умственным развитием. К сожалению, и по отношению к этому заболеванию из группы врожденных системных мы вынуждены сказать, что научно обоснованной профилактики и лечения в нашем распоряжении пока еще не имеется. Специального упоминания требует одна вполне определенная разновидность остеохондродистрофии, которая вызывает большой интерес в литературе последнего времени. Это синдром, или болезнь Пфаундлера — Гурлер, или так называемый гаргойлизм (gargoylismus от старо-французского слова gargouille, что означает выступающий наконечник водосточной трубы, в виде фантастической фигуры), или липоостеохондродистрофия.
Гаргойлизм — это особый тип остеохондродистрофии раннего детского возраста, который отличается не только некоторыми второстепенными деталями костных изменений, сколько сочетанием костных изменений с резко выраженной клинической картиной поражения многих важных систем в противовес более или менее нормальному общему развитию больных, страдающих неосложненной остеохондродистрофией. Всегда резко выражены психические изменения, умственная отсталость и неполноценность в отличие от основной формы остеохондродистрофии, когда интеллект остается нормальным. Довольно меткое название гаргойлизм оправдано весьма типичным внешним обликом ребенка, действительно крайне уродливого, напоминающего средневековую химеру, страшилище, не похожего на обычного кретина. Гаргойлизм — это не только всегда карликовый рост, но и непропорционально крупная голова с обезображенными чертами лица. Глаза широко расставлены (так называемый гипертелоризм), часто имеется косоглазие; как правило, налицо помутнение роговиц. Слух понижен. Нос имеет седловидную крючковатую форму, его спинка придавлена. Из носа сочится густое и обильное гнойное отделяемое. Надбровные дуги сильно выпячены вперед, нависают над глазами. Верхняя челюсть мала, недоразвита, а нижняя широка и удлинена, она выступает вперед (прогнатия). Кожа суха. Волосы коротки, жестки и колючи. Брови удивительно густые, пушистые. Печень и селезенка заметно увеличены. Позвоночник всегда поражен. Шея особенно коротка и толста. Искривление позвоночника кзади в виде прямого угла — постоянный и обязательный симптом, так что уже маленькие больные дети, когда они способны встать на ножки, сильно скрючены. Часты грыжи — пупочная, паховые. Обязательно деформированы нижние конечности — колени и стопы вальгированы, пальчики рук и ног скрючены, подвижность конечностей в суставах ограничена, дети обычно плашмя лежат. Рентгенологическая картина — это, помимо всего, резко усиленная степень остеохондродистрофии. Если, однако, при чистой форме остеохондродистрофии череп грубых изменений не представляет, то при гаргойлизме он является долихо- или оксицефалическим. Гипофизарная ямка соответственно удлинена и уплощена, клиновидные отросточки плохо развиты. При общей тяжелой пластиспондилии недоразвитие передних половинок тел позвонков и характерная ступенчатость захватывает обычно и XII грудной позвонок, и I и II поясничные позвонки. Эта деформация тела или тел позвонков для гаргойлизма обязательна. На рентгенограмме таза видны плоские вертлужные впадины, а деформация верхних концов бедренных костей имеет, вероятно, в связи с адинамией больных маленьких детей, не варусный, а вальгусный характер, т. е. шейки бедер симметрично с обеих сторон толсты и длинны, эпифизарные головки плоски, расширены, увеличены. Отметим еще уплощение лопаток, утолщение грудинных концов ключиц, неуклюжие большие головки плечевых костей, а иногда и утолщение средних отделов диафизов больших трубчатых костей. Утолщены на всем протяжении и ребра, только их шейки истончены. Процесс окостенения совершается с резким запаздыванием. Гаргойлизм обычно ведет к ранней смерти. При посмертном исследовании обнаруживаются изменения со стороны многих систем организма, вызванные каким-то очень глубоким нарушением обмена веществ. Вероятнее всего, что в основе этого своеобразного, но еще недостаточно изученного заболевания стоит процесс отложения в тканях и клетках какого-то не свойственного нормальному организму вещества, дающего гистохимически реакцию па гликоген. Этот гликоген может быть связан с белковыми телами. Отсюда универсализм поражений. Имеется поражение центральной нервной системы, а также ретикуло-эндотелия, сердечно-сосудистого аппарата, эндокринной и костной систем. Чужеродное вещество, откладывающееся в мозгу, сперва склонны были отнести к липоидам. Но оказалось, что гаргойлизм не имеет ничего общего с липоидными заболеваниями (ксантоматозом, болезнью Гоше, амавротической идиотией Тей-Закса и т. д.), так что название липоостеодистрофия следует считать неоправданным. Все попытки оказать больным лечебную помощь, в том числе при помощи эндокринных препаратов и измененной диеты, пока оставались без всякого успеха.
5. МНОЖЕСТВЕННЫЕ ЭПИФИЗАРНЫЕ ДИСПЛАЗИИ
Множественные эпифизарные дисплазии описаны впервые бесспорно Мурком Янсеном (Murk Jansen) в 1934 г., однако их вполне обоснованно выделил в самостоятельную нозологическую единицу в 1935 г. Томас Фербенк (Thomas Fairbank), который предложил общепринятое в настоящее время название — множественные эпифизарные дисплазии. Это также чаще всего семейно-наследственная болезнь, однако приходится наблюдать и спорадические случаи заболевания. Эти дисплазии начинаются всегда в детстве. Внешние проявления сказываются медленно, исподволь, так что у юношей и взрослых невозможно точно установить время клинического начала. Основные жалобы больных сводятся к некоторой низкорослости, но не карликовости, причем общие отношения длины туловища к длине конечностей остаются в основном пропорциональными. Беспокоят также, кроме косметической стороны, боли в конечностях, тугоподвижность в суставах, изменения походки и иногда главным образом развивающиеся деформации конечностей, в первую очередь нижних. Оба пола одинаково подвержены. Интеллект остается нормальным. Эндокринных нарушений заведомо не имеется. Множественные эпифизарные дисплазии представляют собой своеобразное системное нарушение процессов энхондрального окостенения. Наиболее „ответственные” ядра окостенения, т. е. ядра окостенения в области крупных суставов — тазобедренного, коленного, плечевого и лучезапястного, но не локтевого — появляются с опозданием, развиваются замедленно и неправильно, слияние эпифизов с метафизами также осуществляется неправильно в запоздалые сроки. При гистологическом исследовании каких-нибудь специфических патологических процессов не обнаруживается, имеется лишь нарушение стройности энхондрального костеобразования. Этиология этого врожденного заболевания остается неустановленной. Рентгенологическое исследование имеет важное практическое значение, так как только оно обеспечивает безупречное распознавание. При незнакомстве рентгенолога с множественными эпифизарными дисплазиями неизбежны диагностические ошибки. Наш опыт также учит тому, что множественные эпифизарные дисплазии — это отнюдь не редкая болезнь, многие случаи заболевания, особенно нерезко выраженные, остаются просто нераспознанными, не названными своим именем.
{kind=link}
Рентгенограмма области обоих коленных суставов. Уплощение эпифизов, особенно большеберцовых костей. Плоская межмыщелковая ямка в дистальном эпифизе бедренной кости с обеих сторон. Этому способствует еще малая обращаемость больных, когда они не испытывают каких-нибудь значительных неудобств, как и другие члены их семей. Сплошь и рядом диагноз ставится только в результате рентгенологического исследования, предпринятого по другому поводу. Чаще всего больные попадают к нам по назначению эндокринологов, озадаченных отсутствием у больных определенных клинических симптомов эндокринопатии. Рентгенологическая картина весьма характерна, и рентгенологическое распознавание теперь трудностей не представляет. Необходимо, однако, иметь в виду, что эта болезнь отличается довольно значительным многообразием в отдельных спорадических случаях и в разных семьях, в то время как в пределах одной и той же семьи проявления, конечно, с учетом возрастного фактора, почти одинаковы. Важно, что при множественных эпифизарных дисплазиях все изменения в конечностях симметричны. Наиболее показательны патологические сдвиги в области обоих коленных (рис. 284) и лучезапястных суставов. Энхондральные линии неправильны по ходу, изогнуты, иногда змеевидны. Эпифизарные ядра уменьшены в размерах, лишены нормальной формы, угловаты и полигональны, их структурный рисунок нарушен, иногда пятнист, они окаймлены неровными зазубренными контурами. У малых детей ядра расщеплены на несколько фрагментов.
{kind=link}
Оба голеностопных сустава. На контрольных рентгенограммах позвоночника высота позвонков нормальна, брахиспондилии или платиспондилии нет. Центральная часть дистальных эпифизов лучевой и локтевой костей сильно вдавлена, и эпифизы приобретают форму буквы V. В них вдавлены уменьшенные и неуклюжие кости запястья. Сильно расплющивается с обеих сторон головка плечевой кости. Головка бедренной кости справа и слева также деформирована, уплощена, угловата, может фрагментироваться, а в редких случаях и полностью отсутствовать. Дистальные мыщелки бедра резко уплощены, расплющены, прямоугольны, так что межмыщелковая ямка неглубока, поверхностна, плоска, почти отсутствует. Рентгеновские суставные щели повсеместно неправильны, то сужены, то расширены, то для примера в пределах одного коленного сустава различны в медиальной и латеральной частях. Нарушение нормальной картины эпифизов особенно отчетливо выражено и в дистальных эпифизах большеберцовых костей (рис. 285), которые резко истончены, и ось конечности отклоняется в сторону. Значительное участие принимают в картине болезни и ядра окостенения в костях предплюсны и запястья, так что множественные эпифизарные дисплазии, вопреки их названию, — это процесс не только в эпифизах, но и в коротких губчатых костях. Все это ведет к укорочению конечностей, но укорочение рук и ног не достигает значительной степени и во всяком случае, как уже выше сказано, не нарушаются пропорции тела. Деформации выражаются чаще всего в незначительной степени варусной или вальгусной деформации бедер, коленного или голеностопного суставов, плеча, нередко в плоскостопии. В дальнейшем у взрослых больных рано наступают закономерные вторичные изменения типа деформирующего остеоартроза. Диафизы при множественных эпифизарных дисплазиях остаются нормальными, они разве только немного изогнуты вторично. Равным образом не втягиваются в патологический процесс и метафизы — они только немного булавовидно вздуты, также вторично, вслед за увеличением поперечника эпифизов. Подчеркнем, что рентгенологически череп и позвоночник никаких существенных уклонений от нормы не представляют. Не поражены челюсти и зубы. Лишь весьма незначительно или вовсе не изменены пястные, плюсневые кости и фаланги пальцев рук и ног. Рентгенологическое исследование имеет в большинстве случаев заболевания решающее дифференциально-диагностическое значение. Первое место занимает здесь остеохондродистрофия. Нетрудно отметить, что клинико-рентгенологическая картина при обоих заболеваниях во всем сходна. Разграничивает же и ставит обе болезни в противоположные полюса состояние позвоночника: он остается при множественных эпифизарных дисплазиях нормальным и совершенно обязательно представляет едва ли не самые важные в диагностическом отношении изменения при остеохондродистрофии. И опять-таки не случайно в литературе возникают указания, что множественные эпифизарные дисплазии — это „атипическая остеохондродистрофия”, „атипическая болезнь Моркио”, „болезнь Моркио с нормальным позвоночником” и т. д. Таковы же соображения и относительно дифференциальной диагностики с гаргойлизмом и хондродистрофией. И мы лишний раз убеждаемся в том, что все эти „атипические” „варианты” хондродистрофии и остеохондродистрофии — это лишь нераспознанные, непонятые из-за неосведомленности различные, но в настоящее время уже четко выделенные самостоятельные нозологические единицы, которые рентгенолог обязан четко дифференцировать. Очень важно, да и не трудно, как мы это нередко видим в практике, не допускать смешения множественных эпифизарных дисплазий с гипотиреозом. На основании совокупности клинических и рентгенологических данных дифференциальная диагностика не сложна. В одном нашем случае нормальная функция щитовидной железы была доказана при помощи радиоактивного йода. Гормональное лечение при гипотиреозе и кретинизме дает отличные результаты, между тем как при дисплазиях оно неэффективно и бессмысленно. Как при остеохондродистрофии, так и при эпифизарных дисплазиях изолированная картина таза может стать причиной грубой диагностической ошибки — необоснованного диагноза болезни Легг—Кальве— Пертеса. Контрольные снимки области коленных и лучезапястных суставов должны своей положительной и богатой рентгенологической находкой стать основанием для правильного диагноза системного врожденного заболевания. Для остеохондропатии также характерно строго закономерное фазовое течение, чего нет при множественных эпифизарных дисплазиях. Много общего имеют множественные эпифизарные дисплазии и с точечными эпифизарными дисплазиями. Но это, бесспорно, не одна и та же болезнь. Решающее дифференциально-диагностическое значение имеет, помимо тонкостей рентгенологической картины, возрастной фактор: точечная дисплазия — это болезнь плода, новорожденного и ребенка в первые месяцы его жизни, в то время как множественные эпифизарные дисплазии получают свое клинико-рентгенологическое значение только в ясельном и школьном возрасте. Различны течение и исходы обеих болезней. Ряд схожих черт объединяет рентгенологические проявления при врожденных эпифизарных дисплазиях и болезни Кашина—Бека, но дифференциация проста хотя бы на основе географических и эндемических соображений. Что касается лечения и предсказания, то влиять на болезненно протекающий энхондральный процесс мы еще не умеем. Существенное улучшение в смысле общего развития скелета может быть получено благодаря физио- и бальнеотерапии. Общее предсказание при множественных эпифизарных дисплазиях в основном благоприятное, болезнь ведь особенно грубых деформаций и во всяком случае калечения не вызывает. При более значительных варусных и особенно вальгусных деформациях коленного и голеностопного суставов показаны в позднем юношеском возрасте и предпочтительно у взрослых исправляющие ортопедические операции, те или иные остеотомии. Важно воздержаться от научно непоказанных гормональных препаратов, в первую очередь от препаратов щитовидной железы. Особую разновидность эпифизарных дисплазий составляет односторонняя эпифизарная дисплазия — dysplasia epiphysaria hemimelica. Она также называется тарзомегалией, или еще тарзо-эпифизарной аклазией. Впервые эта врожденная болезнь описана в 1926 г. французскими рентгенологами Муше и Бело (Mouchet et Belot), но особенно подробно изучена также Фербенком. В литературе описано около полсотни казуистических наблюдений. При этой болезни неправильность развития происходит у детей в возрасте 2—8 лет при определенных условиях, а именно в дистальных эпифизах больших трубчатых костей одной только нижней конечности, односторонне, асимметрично. Поражаются главным образом дистальный эпифиз большеберцовой кости и почему-то излюбленно таранная кость. Эти участки скелета разбиваются на ряд крупных фрагментов, с неправильной структурой, подчас симулирующей из-за наличия участков затемнения асептический некроз, расчлененные ядра окостенения имеют угловатую неуклюжую форму. Процесс костного развития происходит эксцентрично — одна половина эпифиза чрезмерно велика, другая мала, и поэтому возникают осевые отклонения в сторону, как правило, стопа отодвигается в латеральном направлении, т. е. деформация имеет вальгусный характер. Диафиз и метафиз по существу не меняются, или же только вторично несколько перестраиваются. Клинически налицо костная припухлость, ошибочно нередко принимаемая за костно-хрящевую опухоль. Хорошие результаты дает хирургическое вмешательство — иссечение разросшейся лодыжки и исправление деформативного процесса в суставе.
6. МНОЖЕСТВЕННЫЕ МЕТАФИЗАРНЫЕ ДИСПЛАЗИИ (СИНДРОМ ПАЙЛЯ)
Эта врожденная системная болезнь скелета — одна из наиболее редких из всех дисплазий. Она весьма характерна с клинико-рентгенологической точки зрения. Ее распознавание целиком зависит от компетенции рентгенолога и при знакомстве с этой нозологической формой не составляет труда. Эта дисплазия четко описана впервые в 1931 г. Пайлем (Pyle), хотя и до этого автора были опубликованы единичные казуистические наблюдения. Это болезнь детского возраста с определенным семейным характером. Клинически на первый план выступают вальгусные коленные суставы, невозможность разогнуть предплечья в локтевых суставах, утолщения больших трубчатых костей, непропорционально длинные ноги. Как правило, налицо значительные изменения черепа — мозгового и лицевого, и ряд авторов предпочитает по этой причине назвать болезнь черепно-метафизарными дисплазиями (семейными кранио-метафизарными дисплазиями). У больных большая голова, широко расставленные глаза (гипертелоризм), небольшой плоский седловидный нос с широкой (запавшей) спинкой, обязательно резко отстающие в развитии плохие, крошащиеся зубы. При таких изменениях черепа непременно следует проверить рентгенологически скелет конечностей, иначе точный диагноз может и не быть поставлен. Все биохимические показатели нормальны. Рентгенологически определяется яркая картина симметричных значительных бутылообразных или булавовидных вздутий не только метафизов больших трубчатых костей, но собственно и соответствующих третей и диафизов, а средняя часть диафизов остается не расширенной. Корковое вещество на местах вздутий истончено. Имеются и небольшие осевые искривления костей. Больше всего поражены концы костей, образующих коленный сустав. Вздуты таким же образом и медиальные половины ключиц, подчас также передние концы ребер. Позвоночник остается нормальным. Череп, помимо уже клинически определяемых изменений, показывает на рентгенограммах симметричный гиперостоз покровных костей свода и челюстей. Все придаточные полости носа содержат не воздух, а заполнены костными разрастаниями. Течение этой болезни довольно благоприятное, лишь мало и медленно прогрессирующее. Оно омрачается главным образом сравнительно частыми тяжелыми по своим последствиям сужениями костных отверстий и каналов черепномозговых нервов и сосудов. Дифференциальная диагностика синдрома Пайля не проста. Она требует учета довольно большого ряда различных болезней, в первую очередь черепно-ключичного дизостоза, болезни Гоше, лейкоза, эритробластоза и в особенности фиброзной дисплазии черепа и леонтиаза.
7. ВРОЖДЕННАЯ ЭПИФИЗАРНАЯ ТОЧЕЧНАЯ ДИСПЛАЗИЯ
В последние годы выделена еще одна сравнительно редкая врожденная болезнь—эпифизарная точечная дисплазия скелета (dysplasia epiphisaria punctata congenita). Мы давно, еще в 20-х годах, наблюдали несколько случаев этого заболевания и в полном согласии с Фербенком (Fairbank) безоговорочно считаем ее самостоятельной нозологической формой. Врожденная эпифизарная точечная дисплазия представляет немалый педиатрический рентгенологический интерес. Описано несколько случаев внутриутробного поражения. Практически же — это болезнь раннего детского возраста, она обнаружена пока только у маленьких детей, притом спорадически, без семейного проявления. Чаще она наблюдается у девочек, чем у мальчиков. Сказывается эта болезнь рентгенологически в том, что (см. рис. 261, М) вместо нормальных правильных одиночных ядер окостенения у развивающегося ребенка в области эпифизов появляется множество собранных в кучку мелких точечных костных образований. Нормальное ядрышко окостенения стало быть распадается, распыляется на целый комплекс ядрышек, занимающих более обширное место в эпифизарном конце кости. Эти точечки исключительно плотны, бесструктурны. Они имеют округлую форму и различную величину. Появляются они обычно значительно раньше положенных сроков, иногда опережающих контрольную возрастную группу на несколько лет. Точечные ядрышки способны в дальнейшем по числу и размерам уменьшаться, они могут рассасываться, но иногда, наоборот, они увеличиваются и сливаются друг с другом. Поражены при этом заболевании именно эпифизы, в меньшей степени апофизы, и при дальнейшем формировании скелета эпифизы становятся крупными, неуклюжими. Эпифизарные ростковые линии теряют свой прямой ход, они искривляются, волнисты, расщеплены. Рост костей в длину довольно значительно тормозится, и трубчатые кости остаются подчас сильно укороченными, что может повлечь за собой ту или иную степень карликовости. Весь этот процесс в эпифизах происходит симметрично. Наиболее часто и резко поражаются эпифизы бедренной кости, проксимальные концы обеих большеберцовых костей (значит, особенно показательны рентгенограммы области коленного сустава), проксимальные эпифизы плечевых костей, а дистальные эпифизы большеберцовых костей, оба конца малоберцовых костей, дистальные концы костей предплечья и скелеты кистей и стоп вовлекаются в болезненный процесс меньше всего. Важно, что кроме костных поражений имеются и другие, притом глубокие системные изменения. Больные дети обычно обнаруживают при врожденной эпифизарной точечной дисплазии общую слабость, умственную отсталость, часто наблюдаются катаракты, утолщение кожных покровов, имеется тугоподвижность суставов, позднее вставание на ножки и затруднения при ходьбе. Этой болезни свойственна высокая общая летальность еще в младенческий период жизни. Дифференцировать редкую врожденную эпифизарную точечную дисплазию приходится прежде всего с кретинизмом, который также обусловливает нарушения энхондрального развития костей. Важное отличительное распознавание построено на отсутствии при дисплазии в клинической картине признаков гипотиреоза и вообще эндокринных нарушений, а рентгенологически ядра окостенения появляются значительно раньше, а вовсе не позже нормальных сроков. Наконец, остеопойкилия исключается уверенно на том основании, что эпифизарная дисплазия — это поражение, строго ограничивающееся одними только эпифизами и апофизами — и только, в то время как остеопойкилия представляет собой пятнистый процесс в эпифизах и метафизах без срыва всего нормального энхондрального развития скелета. Локализация поражений в скелете строго различна — наибольшие изменения при эпифизарной дисплазии гнездятся в области коленных суставов, тогда как при остеопойкилии они располагаются более периферически, например, в области кистей и стоп. Совершенно различными являются и возраст больных. И самое главное расхождение заключается в диаметрально противоположном клиническом фоне, на котором разыгрывается рентгенологическая картина при обоих врожденных заболеваниях. Если при остеопойкилии клиники, можно сказать, по существу и нет, то при эпифизарной точечной дисплазии симптоматология, течение и исход заболевания весьма серьезны и неблагоприятны. Специальных комментариев требует одна редкая врожденная системная болезнь, впервые выделенная в 1914 г. Конради (Conradi) и получившая название врожденной кальцифицирующей хондродистрофии (chondrodys-trophia calcificans congenita s. punctata, dysplasia epiphysalis punctata, chondroan-giopathia calcarea s. punctata и пр.). Она является своеобразной переходной формой между эпифизарными дисплазиями и хондродистрофией. И все же название „хондродистрофия”, с нашей точки зрения, дает повод к недоразумениям и его следует считать неудачным. Между обеими болезнями имеются некоторые черты сходства, но, бесспорно, это опять-таки не разновидность хондродистрофии или атипическая форма ее, а скорее всего она должна быть включена в круг именно точечной дисплазии. Эта болезнь может быть семейной или спорадической. Девочки заболевают несколько чаще, чем мальчики. Обычно это случайная находка при рентгенологическом исследовании по другому поводу, у новорожденного или грудного младенца, который не обнаруживает никаких клинических, эндокринных, биохимических и других уклонений от нормы, или же все же сочетается с другими неправильностями развития других органов или систем. Рентгенологически определяется множество симметричных, но не зеркально одинаковых мелких пятнистых известковых включений в эпифизарных хрящах трубчатых костей, в коротких костях и позвонках. Конечности обычно или вовсе не укорочены, или же укорочены в небольшой степени, чуть утолщены и искривлены, причем это касается только бедер и плечей. Важно, что „распад” ядер энхондрального окостенения самостоятельно ликвидируется в первые 3—5 лет жизни.
8. ВРОЖДЕННЫЕ СИСТЕМНЫЕ ДИАФИЗАРНЫЕ ГИПЕРОСТОЗЫ
Врожденные системные диафизарные гиперостозы (синонимы: генерализованный гиперостоз и эностоз, врожденная множественная гиперостотическая болезнь, множественные диафизарные склерозы или склерозирующие гиперостозы и т. д.) — это известная еще в старое дорентгенологическое время болезнь, которая в свете современных клинико-рентгенологических данных безоговорочно заслуживает быть выделенной в самостоятельную нозологическую форму. С новых рентгенологических позиций эта болезнь описана сотрудником Роберта Кинбека Энгельманом (Engelmann) в Вене в 1929 г. и в литературе нередко без особых к тому оснований обозначается как болезнь Энгельмана, иногда и Камурати—Энгельмана (Camurati). Это заболевание, несомненно, одно из наиболее редких представителей всей группы врожденных системных заболеваний скелета. Оно встречается как в спорадическом, так и в явно семейном выражении. Правильно указывают Д. Г. Рохлин и М. А. Финкельштейн, что обычно при исследовании подобных больных рентгенологи не интересуются состоянием костно-суставного аппарата более молодых членов семьи и что при этом надо полагаться не на опрос родителей и не на результаты клинического исследования, так как, вопреки отрицательным данным, полученным таким путем, рентгенологическое исследование позволяет выявить значительные распространенные изменения скелета. Из общеклинической характеристики страдающих врожденными системными диафизарными гиперостозами следует выделить то общее, что эта болезнь, будучи несомненно врожденной, проявляется не сразу после рождения ребенка, а лишь значительно позднее. Дети становятся на ножки обычно сравнительно поздно — на третьем — четвертом году жизни — и при этом испытывают затруднения при ходьбе. Во многих случаях первоначальный период жизни ребенка происходит под знаком замедленного развития, дети растут худыми, бледными, плохо прибавляют в весе, но впоследствии это выравнивается. Нередко наблюдается некоторая общая и мышечная слабость. В остальном же больные ничем не отличаются от своих сверстников, имеют нормальный внешний облик. Кровь никаких морфологических и биохимических сдвигов не представляет, в частности нет анемии. Ретикуло-эндотелиальная система нормальна, печень и селезенка не увеличены. Нет никаких эндокринных нарушений. Ясно без лишних слов, что в анамнезе нет никаких обязательных указаний на перенесенные инфекционные болезни, в частности серологические реакции на сифилис отрицательны. Общее предсказание весьма благоприятное. При этих условиях понятно, что развитие диафизарных гиперостозов может растянуться и прогрессировать незаметно, и в ряде случаев болезнь распознается сплошь и рядом только в пубертатном возрасте или даже еще позже. Вероятно, что в каком-то периоде до полового развития или в связи с созреванием наступает приостановка болезни, ее стабилизация. Только при тщательном клиническом осмотре можно отметить, да и то без уверенности, некоторое утолщение конечностей и своеобразную нивелировку поверхности рук и ног, сглаживаются выпуклости и углубления, суставы почти не выделяются, так что конечности кажутся более правильными цилиндрическими, чем у нормального человека. В наших собственных случаях обращал на себя внимание высокий рост больных. Кожа над утолщенными костями может быть натянута, но в остальном не изменена. Наиболее важным в практическом отношении проявлением врожденных гипер-остозов служат боли, которые и побуждают больных раньше или позже обращаться за врачебной помощью. Иногда предъявляются также жалобы на тугоподвижность во всех суставах, на скованность движений. Воли обычно связаны с периодами активации болезни и имеют тогда периодически довольно интенсивный ноющий, глубокий, грызущий характер, которые больные сравнивают с зубной болью. Эти боли усиливаются после физической нагрузки, например в ногах после продолжительной ходьбы.
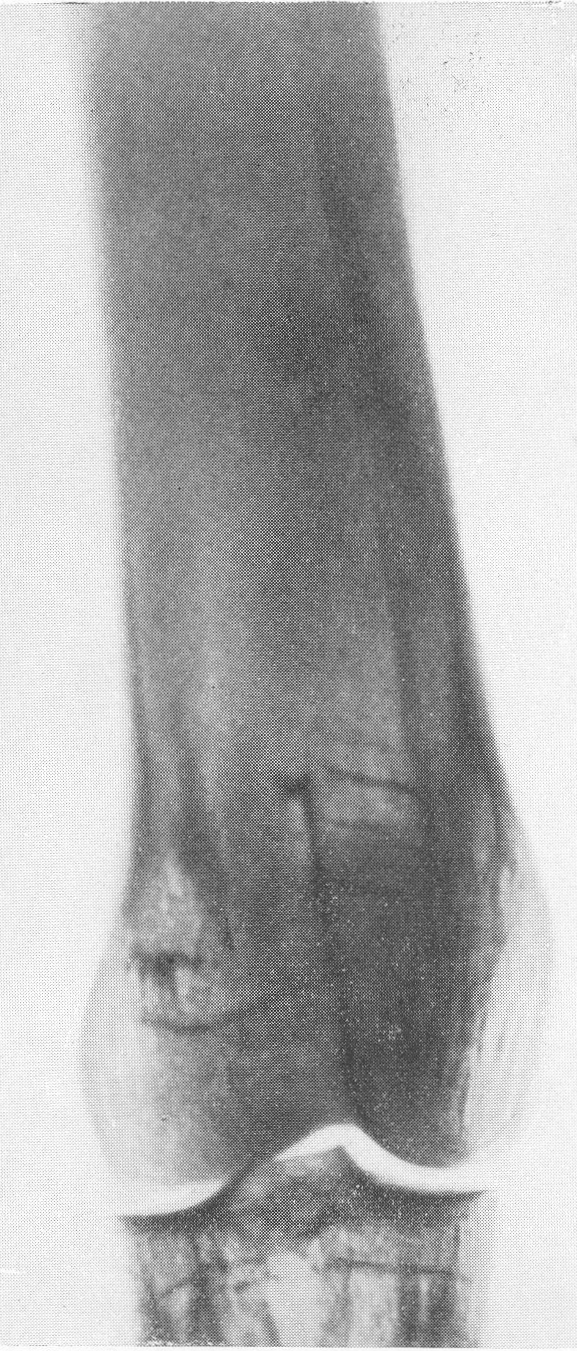{kind=link}
Рис. 286. Врожденные системные диафизарные гиперостозы (болезнь Энгельмана) у 25-летнего мужчины высокого роста. Деталь рентгенограммы дистальной половины бедренной кости и области коленного сустава.
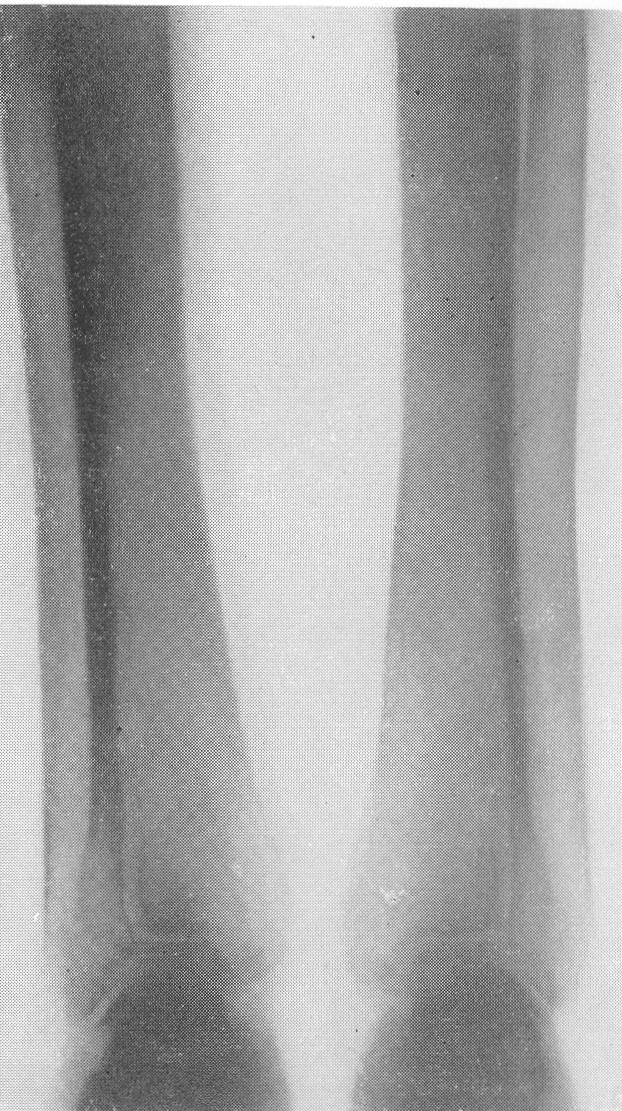{kind=link}
Рис. 287. Врожденные системные диафизарные гиперостозы. 32-летний мужчина патологически высокого роста.
Рентгенограмма обеих голеней. Они определенно относятся к именно диафизам больших трубчатых костей конечностей, а не к суставам. Подчеркнем, что в некоторых случаях, особенно в школьном возрасте, боли могут полностью отсутствовать, и тогда диагноз ставится лишь в результате рентгенологического исследования, предпринятого по другому, не относящемуся к самой болезни или по случайному поводу. При биопсии костной ткани получаются довольно противоречивые заключения. Костная ткань оказывается то нормальной, то обнаруживается утолщение трабекул, то налицо склероз. Заслуживают внимания указания ряда авторов, особенно же Синглтона и сотрудников (Singleton), что болезнь Энгельмана имеет сосудистую основу: под микроскопом определяется системное утолщение стенок сосудов, питающих кости, особенно среднего слоя, с сужением просвета артерий разного калибра, и есть основание приписать роль патогенетического фактора в возникновении остеосклероза редуцированному кровоснабжению костной ткани. Изменения длинных трубчатых костей имеют при врожденных диафизарных гиперостозах самый строгий симметричный характер.Поражаются преимущественно все большие трубчатые кости скелета (см. рис. 261, Н), в меньшей степени малые трубчатые кости. В наиболее сильной степени процесс выражен в бедренных костях (рис. 286), большеберцовых (рис. 287 и 288), плечевых (рис. 289), а в малоберцовой кости, как и в костях предплечья (рис. 290), изменения немного меньше.
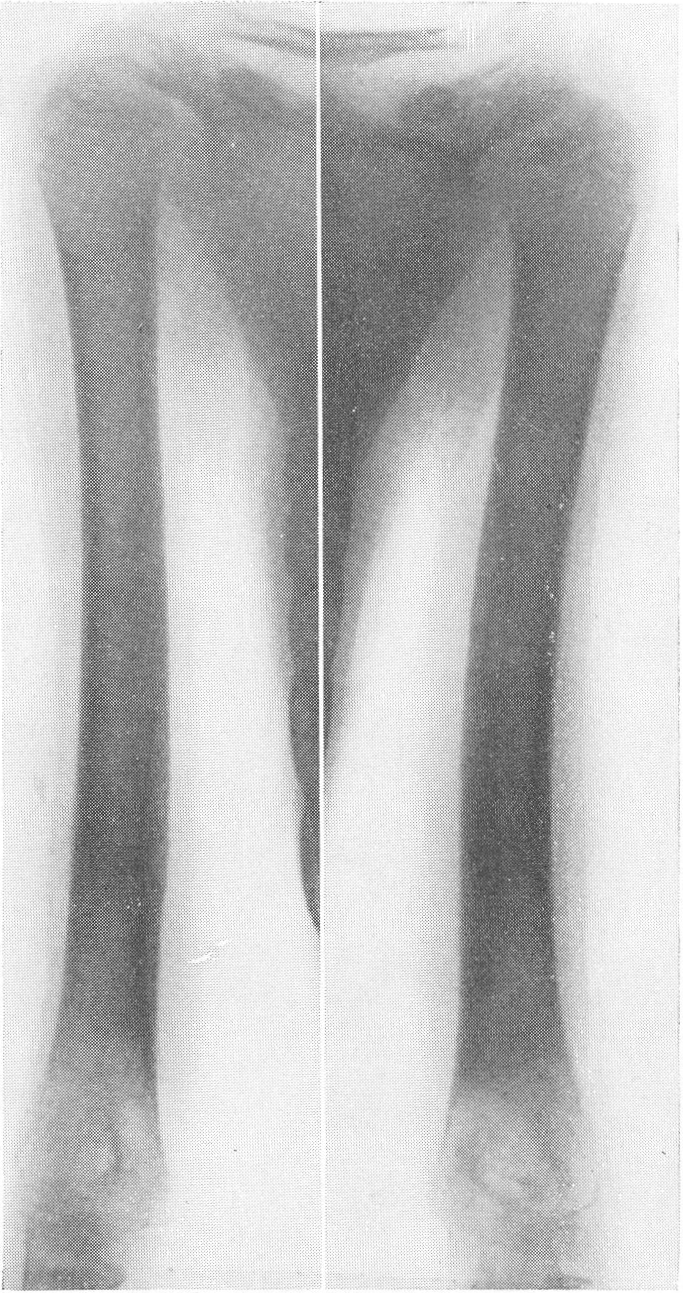{kind=link}
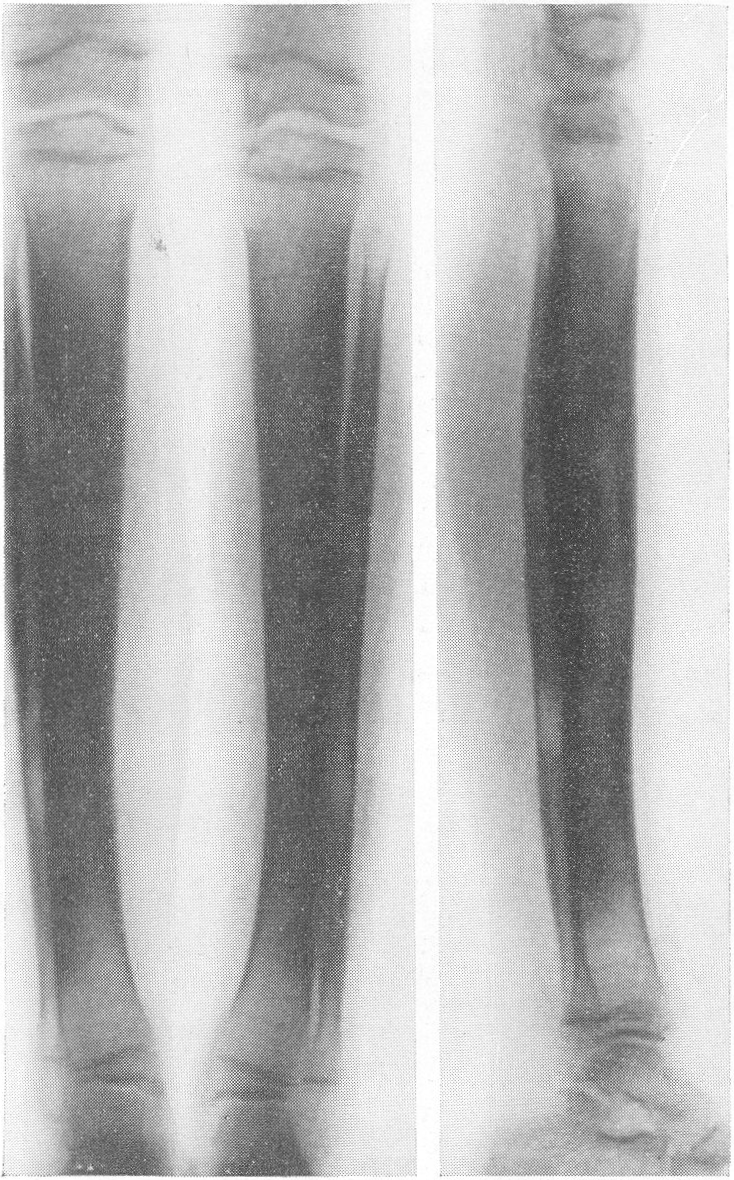{kind=link}
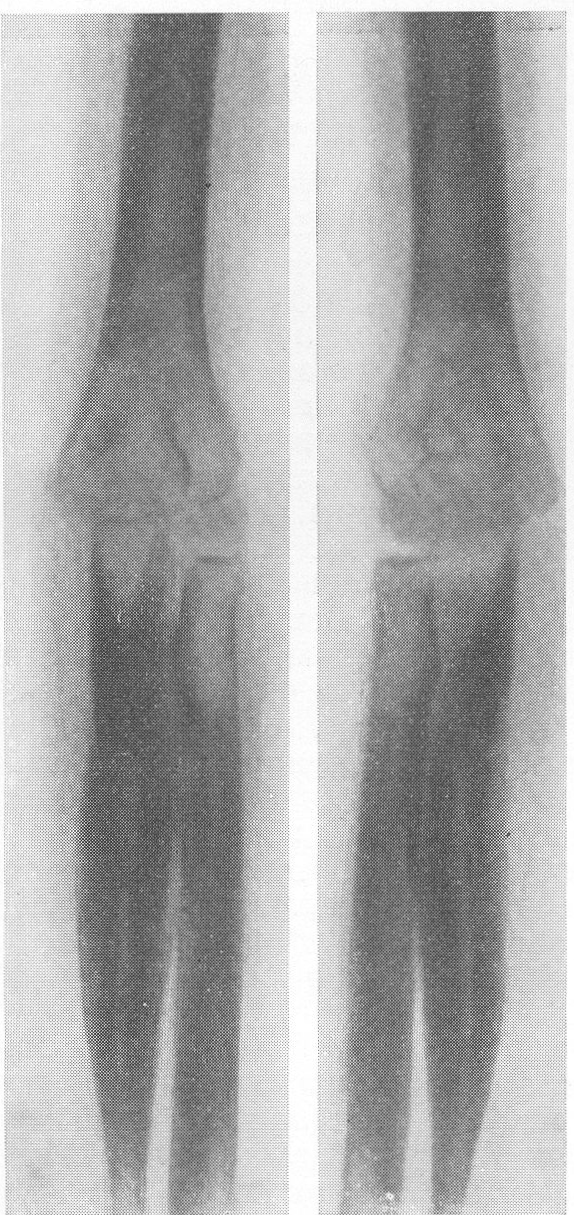{kind=link}
Рентгенограмма обеих плечевых костей и костей предплечья. Структурный же рисунок губчатого вещества типичным образом изменен, а именно многие костные стропила толсты, грубы и весьма плотны, обрывисты. В основном выделяются укрепленные продольные балки, но кое-где в эпиметафизах вдруг резко выступают подчеркнутые подобные же поперечные костные пластины. Все это вполне соответствует, естественно, гистологической картине: биопсия показывает отсутствие каких бы то ни было признаков воспалительного или иного значительного патологического процесса, а имеется лишь некоторый равномерный остеосклероз с узкими гаверсовыми каналами меж уплотненного костного вещества. Груботрабекулярный структурный рисунок при врожденных гиперостозах напоминает губчатое строение при гемангиоме, и сходство особенно велико именно в картине позвонков, притом как в телах, так и в дужках и отростках.
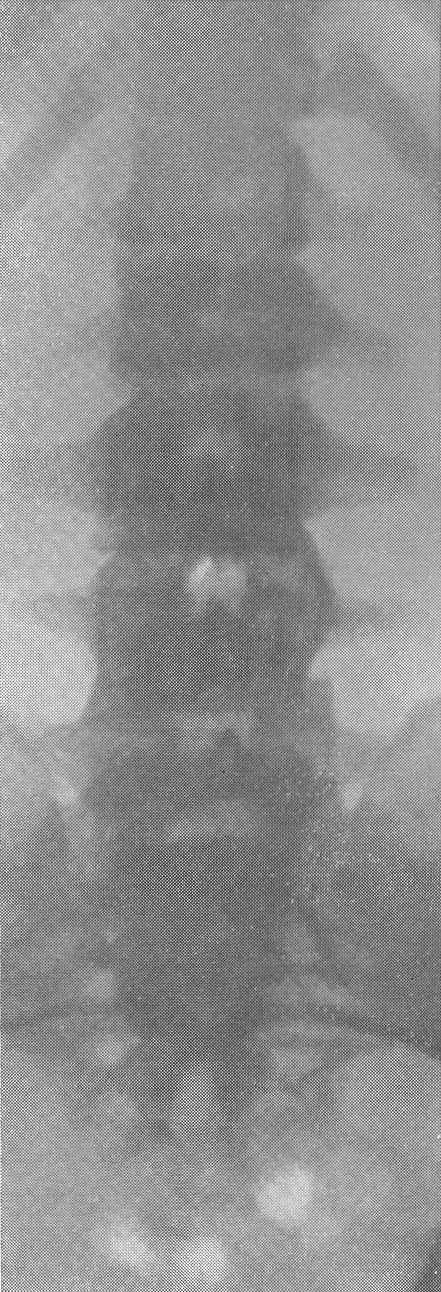{kind=link}
Рис. 291. Врожденные системные диафизарные гиперостозы у того же больного, что на рис. 286. По ясничный отдел позвоночника с характерным структурным рисунком.
Такова рентгенологическая картина в сформировавшихся случаях у взрослых больных. В детском же возрасте структурный рисунок симметрично утолщенных кос гей может быть и вовсе неравномерным, а иметь рассеянный крупнопятнистый очаговый характер, притом в деталях структурной перестройки и не полностью симметричный с обеих сторон (рис. 288) Утолщение костей также может быть не строго цилиндрическим, а несколько веретенообразным. Неоднократно мы, как и другие авторы, наблюдали при врожденных диафизарных гиперостозах некоторый склероз костей основания черепа, в особенности соответственно передней и средней черепным ямкам. Кости свода черепа при этом заболевании неизменно остаются в норме. Из-за недостаточного знакомства с врожденными диафизарными гиперостозами, обусловленного главным образом редкостью заболевания, последнее сплошь и рядом остается нераспознанным, принимается ошибочно за какое-нибудь более известное страдание, и поэтому врожденные гиперостозы приобретают высокое практическое дифференциально-диагностическое значение. Диагностические ошибки здесь особенно нежелательны, так как неминуемо влекут за собой бесполезные, а то и вредные тактические мероприятия при проведении дальнейшей дифференциации, уже не говоря о рекомендации всевозможных лечебных средств, не способных в корне изменить течение врожденного системного заболевания. Учета при отличительном распознавании врожденных гиперостозов требуют главным образом болезнь Педжета, акромегалия, генерализованный гиперпластический периостит, множественные корковые гиперостозы раннего детского возраста, неврофиброматоз и еще некоторые врожденные же системные заболевания скелета. Трудности в отличительном распознавании с деформирующей остеодистрофией Педжета порождаются в первую очередь при заболеваниях в смежных возрастных группах, начиная с Пубертатного, в цветущем возрасте. Доводами в пользу врожденных гиперостозов и против приобретенного обезображивающего процесса могут служить системность, строгая зеркальная симметрия, Правильная равномерность поражений, отсутствие искривлений длинных трубчатых костей и патологических переломов, бес-структурность склерозированного коркового вещества в противовес типичному рентгенологическому педжетовскому рисунку и т. д. Акромегалия, которую с врожденными гиперостозами объединяют высокий рост, неуклюжие длинные и толстые кости, симметричность поражения и другие общие черты, все же легко может быть отвергнута, если строить доказательства на типичном внешнем облике больного акромегалией с обязательными изменениями черепа и всей области гипофизарной ямки, преимущественном поражении костей и стоп, а также фаланг, особенно ногтевых, отсутствии равномерных системных гиперостозов больших трубчатых костей. Генерализованные гиперпластические периоститы отличаются рентгенологически от врожденных диафизарных гиперостозов преимущественным поражением периферических костей — фаланг, пястных и плюсневых костей, костей предплечья и голени и менее всего бедра в противовес врожденному заболеванию. Периостальные наслоения имеют не гемогенную, а сложную структуру, шероховатую поверхность. Как правило, клинически налицо изменения органов грудной полости, виновные в образовании патологического процесса в скелете. Что касается множественных корковых гиперостозов в младенческом возрасте, то надо еще раз (стр. 343) твердо подчеркнуть, что это совершенно различные заболевания, и их смешение с теоретической и с практической точек зрения, как это до сих пор еще многими в литературе делается, совершенно недопустимо. Отличительными чертами служат резкое возрастное различие, особенно также локализация, объем и структура периостальных утолщений при обоих заболеваниях, а также совершенно различный клинический фон, на котором разыгрываются оба столь различных по своей природе заболевания. Неврофиброматоз может вызвать гиперостотические и склеротические изменения в одной или в нескольких костях на одной конечности, которые формально могут иметь большое сходство с рентгенологической картиной при врожденных гиперостозах. Решающим аргументом в пользу врожденного системного скелетного заболевания в спорном случае послужит именно распространенность, системность, двусторонность, да к тому же обязательная симметричность — против ограниченности костных изменений при неврофиброматозе. По этим же линиям проводится также исключение болезни Олье — всегда одностороннего и во всяком случае асимметрического поражения одной конечности, в крайнем случае верхней и нижней конечностей на одной стороне. Те же доводы неотразимы и при мелореостеозе. Что касается однокостной или многокостной фиброзной остеодисплазии, то здесь, помимо всего, ведь корковое вещество истончено, а вовсе не утолщено. Мраморная болезнь проявляется в каждой кости совершенно другими симптомами — локализацией, степенью и характером остеосклероза, а также отсутствием резко выраженных диафизарных утолщений. Наконец, если начать в конкретном случае заболевания почему-нибудь с рентгенологического исследования позвоночного столба, если к тому же ограничиться только снимками этой области скелета (что мы всегда так строго осуждаем!), то можно легко впасть в грубую ошибку, смешав врожденное заболевание с гемангиомой позвонка или позвонков. Обязательное дополнительное исследование костей конечностей, разумеется, внесет ясность в возникший спор.
9. МНОЖЕСТВЕННЫЕ ХРЯЩЕВЫЕ ЭКЗОСТОЗЫ
Множественные хрящевые экзостозы (картилагинарные экзостозы, деформирующая хондродисплазия, диафизарная аклазия) — это отнюдь не очень редкое заболевание. Нам приходится наблюдать не меньше десятка новых больных с множественными экзостозами ежегодно. Этиология заболевания не выяснена. Определенную роль играет семейная передача болезни, и нередко можно проследить ряд поколений с более или менее резко выраженным заболеванием. Мужской пол больше предрасположен к заболеванию, чем женский, отношение числа мужчин к женщинам равно 2—3 : 1. Множественные хрящевые экзостозы лишь в редчайших случаях бывают в прямом смысле этого слова, т. е. непосредственно врожденными. Обыкновенно экзостозы появляются в первые годы жизни, не раньше 6—8-го года жизни, и сильнее всего развиваются в периоде физиологического роста скелета, в детском и юношеском возрасте, чаще всего в периоде от 8 до 18 лет. Вообще-то очень медленный рост экзостозов ускоряется главным образом в периоде полового созревания. С прекращением роста в длину и наступлением синостоза эпифизов с метафизами дальнейшее увеличение экзостозов приостанавливается. Размеры отдельных экзостозов в 3-м и 4-м десятилетии могут даже уменьшиться. Таким образом, рост экзостозов теснейшим образом связан с энхондральным ростом скелета. Поэтому понятно, почему различные нарушения эндокринной корреляции, вызывающие неправильности общего роста в длину, влияют также и на развитие экзостозов. Этиологической связи между эндокринной системой и множественными экзостозами, как это предполагает ряд авторов, на наш взгляд, безусловно не существует. Экзостозы исходят из эпифизарных хрящей. Не исключена, однако, и возможность связи между происхождением множественных хрящевых экзостозов и надкостницей метафизарной области. Существует взгляд, что экзостозы прорастают лишь на местах врожденных дефектов периоста, совпадающих с местами прикрепления какого-нибудь крупного сухожилия. При физиологических условиях периост создает только компактную костную ткань, при патологических же процессах, как, например, при переломах, надкостница может производить и хрящевую ткань. Из костного эпифиза и диафиза экзостозы первично местно никогда не исходят. Экзостоз в своей основе состоит из хрящевой ткани, т. е. в сущности является экхондрозом, и окостеневает подобно нормальной хрящевой ткани. Поэтому и название «хрящевые» экзостозы не совсем точно передает сущность процесса. Эпифизарный хрящ при нормальных условиях всегда создает губчатое костное вещество. Это свойство сохраняется и при патологических условиях, и экзостозы в громадном большинстве случаев имеют довольно правильное губчатое строение. Снаружи спонгиозная ткань заключена в тонкую, но плотную компактную костную скорлупу. Поверхность костного экзостоза, т. е. компактный слой, покрыта гиалиновым хрящом толщиной в несколько миллиметров. Из этой хрящевой головки и происходит дальнейший рост всего экзостоза. Эта хрящевая пластинка не является сплошной, как на суставном конце кости, а местами прерывается, так что костная ткань здесь и там обнажена. Чем ближе к периоду полового созревания и прекращению дальнейшего роста, тем тоньше становится хрящевой покров экзостоза. Наибольшей толщины достигает хрящ при экзостозах в виде кочана цветной капусты. Теперь установлено, что в большинстве случаев над более или менее крупным костным хрящевым наростом развивается и слизистая сумка. Наиболее пышно разрастаются экзостозы близ колена и далеко от локтя. В порядке убывающей частоты экзостозы наблюдаются в области проксимального метафиза большеберцовой кости, дистального метафиза бедра, проксимального малоберцовой кости, затем следуют проксимальный конец плеча и дистальный конец костей предплечья, проксимальный конец бедра, область вертелов, особенно большого, подвздошная кость и лопатка, ключица и т. д. Плюсневые и пястные кости поражаются несколько реже, обычно при большом количестве экзостозов. Редко затронут позвоночник, и тогда экзостозы растут из дужек и оснований отростков, но почти никогда не из тела позвонка. Совсем нередко захвачены передние концы тех или иных ребер. Из периферических костей реже всего поражается почему-то дистальный конец плеча. Кости лицевого и мозгового черепа соединительнотканного перепончатого происхождения никогда не поражаются. Как правило, рост экзостозов происходит симметрично. Недавно развившийся экзостоз непосредственно прилегает к эпифизарной зоне со стороны метафиза, по мере же роста в длину экзостоз все больше и больше отодвигается от эпифизарного конца по направлению к середине диафиза и тогда уже располагается в пределах самого диафиза. Положение экзостоза, таким образом, может быть использовано для приблизительного определения его давности. Вообще-то скорость роста в отдельных случаях весьма индивидуальна и различна, она колеблется в довольно широких пределах. Направление роста и форма экзостозов до известной степени зависит от расположения мышц и сухожилий, а именно отдельные наросты возвышаются над костью в промежутках между плотными сухожилиями и мышечными брюшками, где не сказывается механическое препятствие. Число экзостозов очень широко колеблется — от единицы и десятков до сотен и свыше тысячи. Размеры также резко варьируют — от горошины и еще меньше до величины яблока и головки ребенка. Форма экзостозов бесконечно разнообразна. Отдельные экзостозы больше всего напоминают сталактиты и имеют сходство с шаром, колбой, клювом, шипом и пр. Связь экзостоза с самой костью может быть очень различна. Шаровидные разрастания сидят на поверхности метафиза на более или менее узенькой ножке, или же основание нароста значительно шире, чем его плоская поверхность. Это более часто наблюдаемая так называемая табулярная форма экзостозов. Вытянутые в длину экзостозы, прикрепленные к кости лишь частично, т. е. не всем своим основанием, всегда направлены своей свободной частью к центру кости — в дистальном направлении, если экзостозы сидят на проксимальных метафизах, и проксимально, если они возвышаются на дистальных концах костей. Поверхность экзостозов большей частью гладка или грубобугриста. Рентгенологическое исследование играет при множественных хрящевых экзостозах одну из главнейших практических ролей. Экзостозы могут вызывать чувство напряжения или иррадиирующие боли в суставе. Однако сплошь и рядом медленно увеличивающиеся и не вызывающие болей экзостозы не сопровождаются никакими клиническими симптомами и остаются незаметными как для врача, так и для больного. Ведь больной не знает нормальной анатомии и сплошь и рядом принимает свои костнохрящевые разрастания, так сказать узловатость или суковатость своего скелета, за естественное, нормальное явление. В амбулаторной практике, как показывает наш опыт, обыкновенно допускается все та же элементарная ошибка, которая очень хорошо известна, но тем не менее слишком регулярно повторяется: исследованию подвергается лишь одна область, а не весь скелет, и экзостоз принимается за опухоль, чаще всего за остеому, остеохондрому, нередко сразу же и за саркому. Из множественных экзостозов наиболее часто обращает на себя внимание нарост на медиальной стороне проксимального метафиза большеберцовой кости (рис. 292), и то лишь вследствие сдавления rami infrapatellares nervi sapheni и вызванной этим болезненности. Нередко больного побуждает обращаться к врачу неудобство при ношении обуви, голенище высокого сапога давит на «опухоль». Совсем не редко множественные хрящевые экзостозы „открываются” в результате произведенной впервые рентгеноскопии грудной клетки или при флюорографии по поражениям ребер. Большинство экзостозов недостаточно ясно прощупывается при клиническом исследовании, и истинное представление о числе экзостозов, форме, размерах, положении, развитии, строении и т. д. можно получить, лишь имея перед собой рентгенограммы. Среди всех специалистов именно рентгенологу поэтому и приходится чаще всего наблюдать и изучать множественные хрящевые экзостозы. Все изложенные выше анатомические особенности экзостозов находят свое точное изображение на рентгенограммах — описание их анатомической картины есть в сущности описание и рентгенологической картины (рис. 293). Губчатая структура экзостоза представляет прямое продолжение губчатой сети метафиза в периоде роста или мета-эпифиза после синостоза росткового хряща.
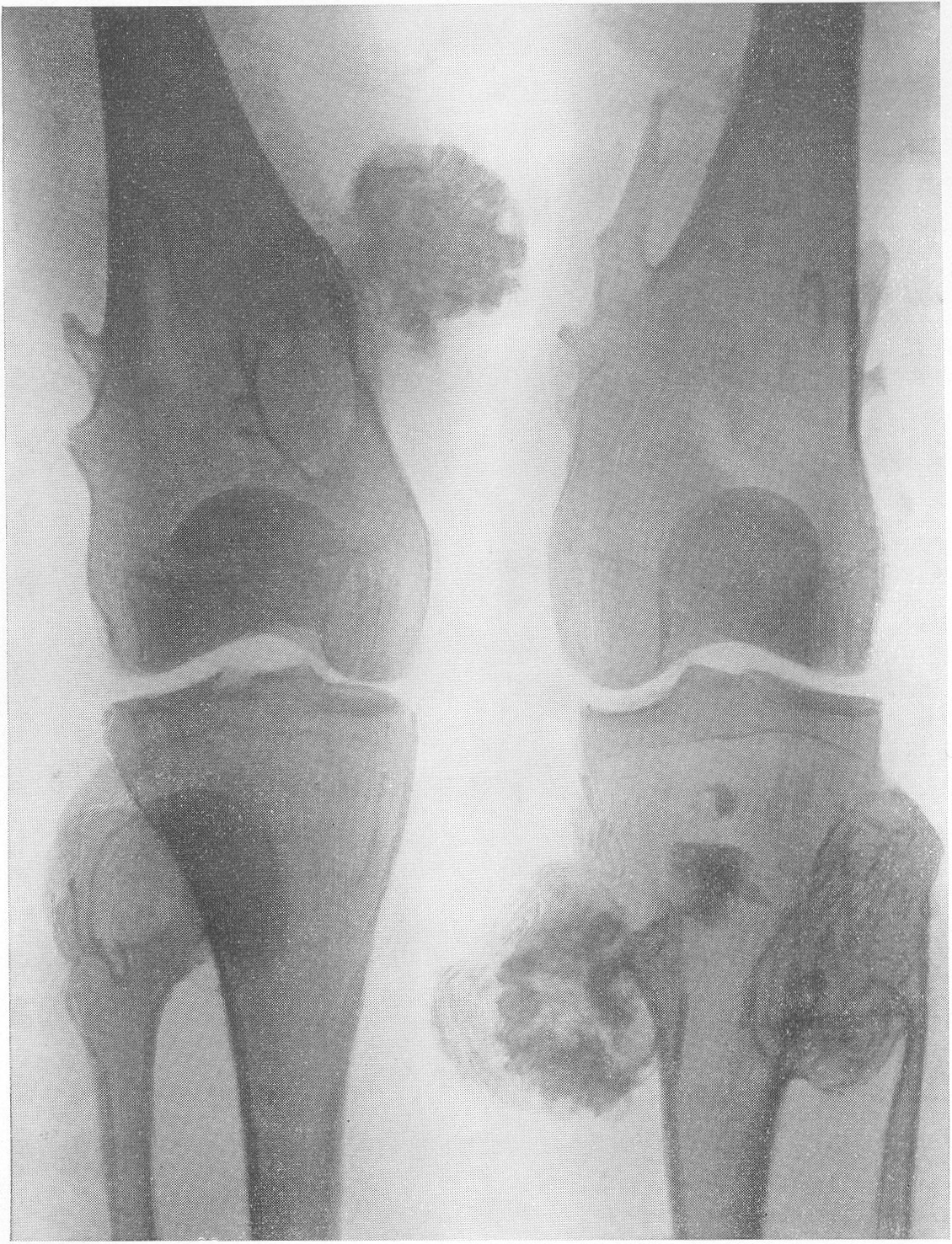{kind=link}
Рентгенограмма области обоих коленных суставов 20-летнего мужчины, обратившегося за врачебной помощью в связи с невозможностью носить высокий сапог на левой ноге. Структурный рисунок экзостоза вполне правилен и совершенен, что собственно противоречит так называемому закону трансформации Вольфа, так как экзостозы не имеют никакого функционального значения. Главные более плотные трабекулы располагаются узким пучком в ножке экзостоза и рассыпаются радиарным букетом к его поверхности. В плоских экзостозах более толстые балочки обыкновенно идут параллельно длиннику кости, или, другими словами, параллельно поверхности экзостоза. И корковое вещество большой цилиндрической кости, постепенно истончаясь, правильной дугой переходит на поверхность экзостоза, покрывая его со всех сторон более или менее тонким слоем компактного вещества. Метафизы, а в периоде после полового созревания и эпифизарные концы утолщены, метафизы расширены и удлинены в сторону диафиза, они становятся неуклюжими, массивными, так что кости представляются характерным образом деформированными. Иногда, в более тяжелых и рано начинающихся случаях, имеется и дугообразное искривление диафизов. Это в особенности касается локтевой и лучевой костей (рис. 294). Локтевая кость в подобных тяжелых случаях также значительно укорочена из-за недоразвития дистального конца, заостренного в виде конуса или наконечника стрелы.
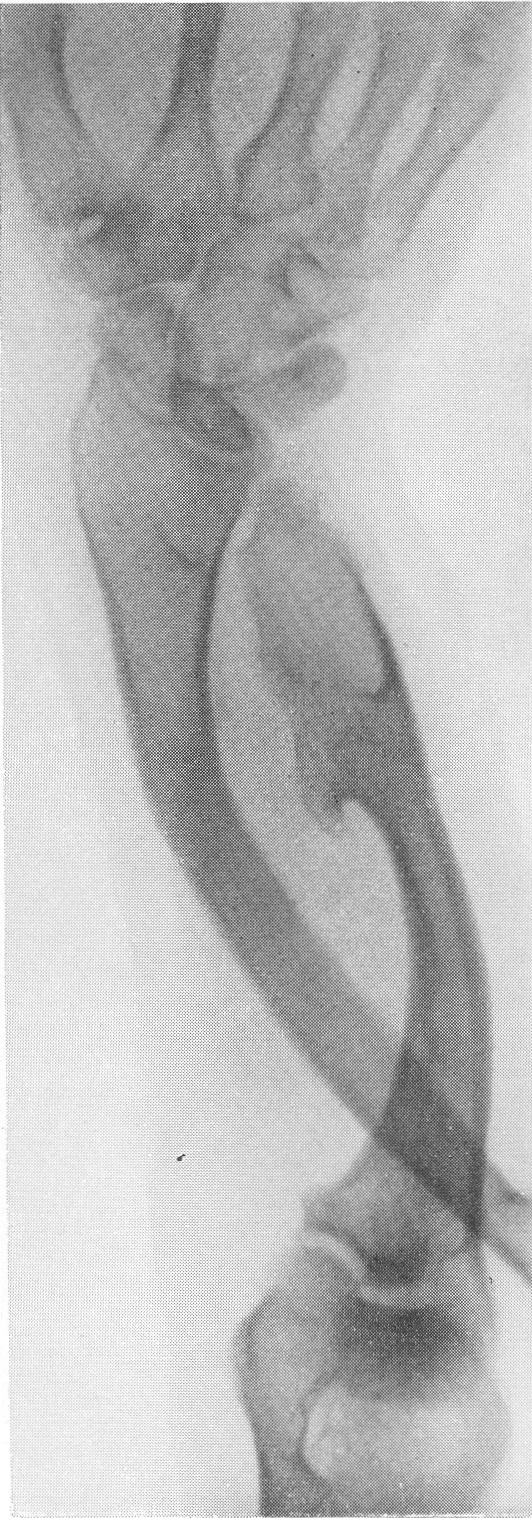{kind=link}
Рис. 294. Множественные хрящевые экзостозы у 34-летнего больного, у которого двое детей с тем же заболеванием. Укорочение локтевой кости и дугообразное искривление обеих костей предплечья. Так называемая симптоматическая маделунговская деформация. Вывих лучевой кости в локтевом суставе.
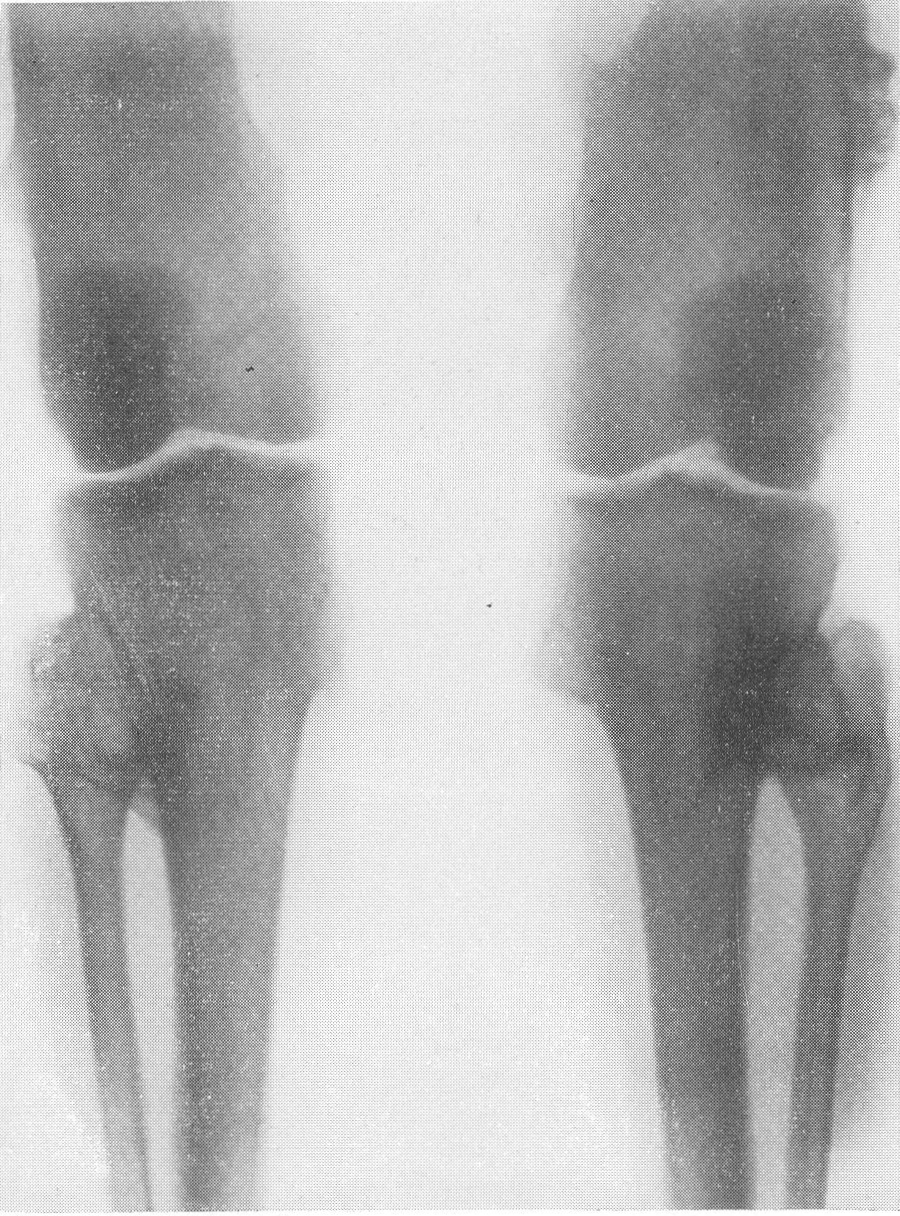{kind=link}
Рис. 293. Множественные хрящевые экзостозы у 25-летнего физкультурника, направленного на рентгенологическое исследование по поводу травматического вывиха левого плеча.
Основное заболевание выявлено лишь рентгенологически. По этой причине при множественных картилагинарных экзостозах и наступает так называемая деформация Маделунга (кн. 2, стр. 213.). В области коленного сустава приходится наблюдать вальгусную деформацию — одностороннюю или двустороннюю, симметричную или по степени выраженности асимметричную. Укорочения и деформации сравнительно часто наблюдаются и в других областях парного или параллельного расположения нескольких костей, например в области пястных и плюсневых костей, и влекут за собой укорочение пальцев и их смещение под углом. Значительное нарушение общего роста в длину, а в редчайших случаях и особый вид карликового роста наблюдаются лишь в очень тяжелых случаях с громадным количеством экзостозов.
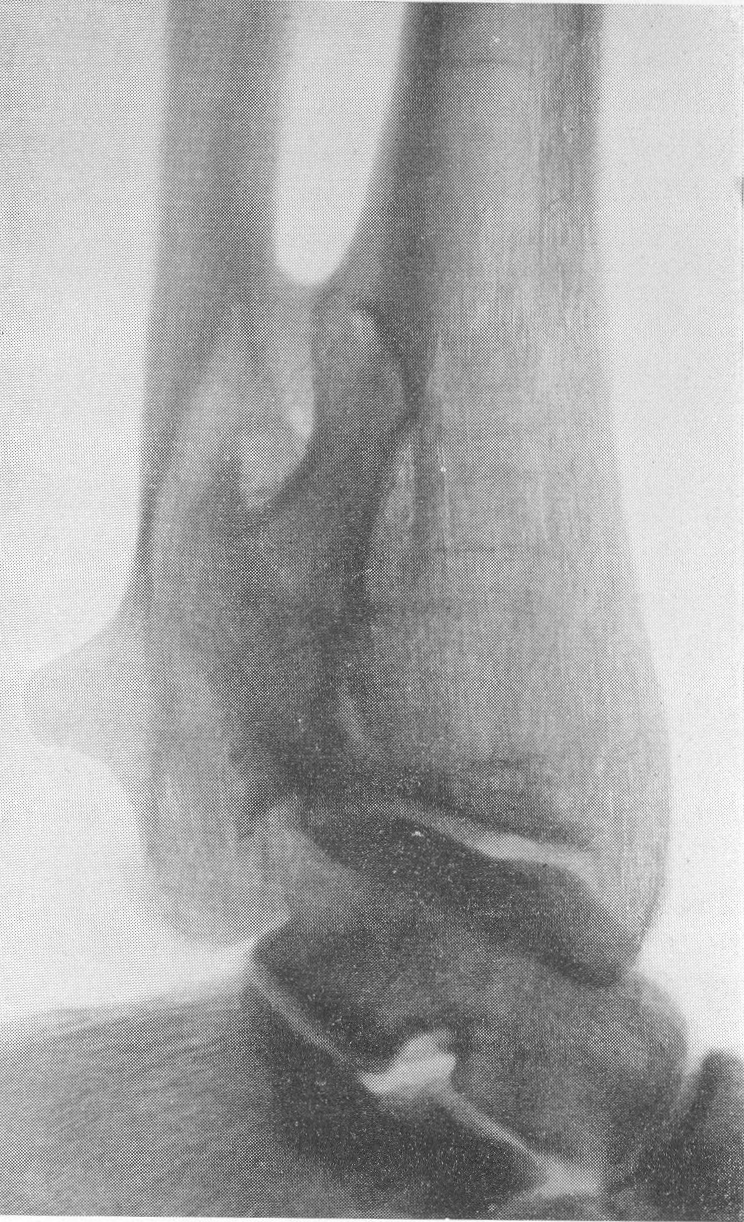{kind=link}
Костное слияние обеих костей голени на протяжении их нижней четверти. Иногда приходится видеть на рентгенограммах и патологические сращения рядом расположенных костей, в первую очередь костей предплечья и голени (рис. 295). Эти костные сращения или слияния имеют вид широкой спайки с правильным структурным рисунком, перекидывающейся через межкостное пространство. Располагается костный мостик, как и все экзостозы, не в средней трети диафиза, а в области метафиза. В других случаях поверхности отдельных экзостозов, лежащих друг против друга, отшлифовываются таким образом, что получается нечто вроде неоартроза — развиваются суставные поверхности, напоминающие выпуклую головку и соответствующую ей по форме вогнутую впадину. Иногда наблюдаются и поверхностные блюдцеобразные изъяны, узуры костей, вызванные давлением экзостоза на соседнюю кость (рис. 296).
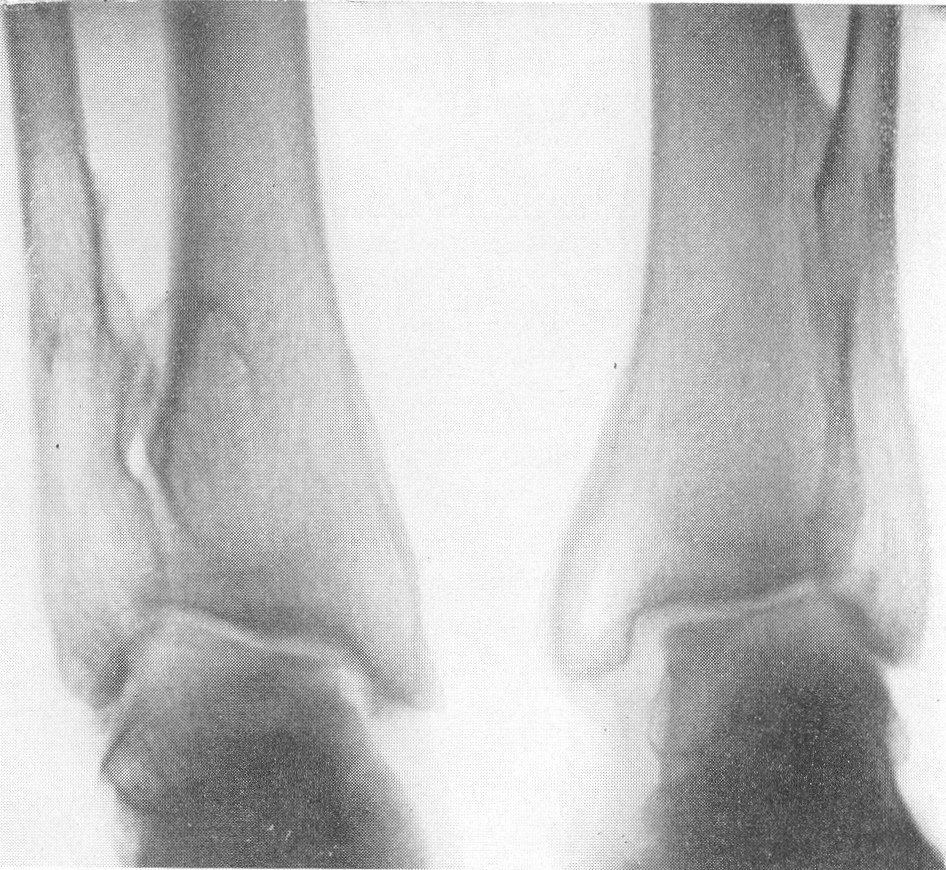{kind=link}
Область нижней половины голени и голеностопных суставов с обеих сторон. Костного слияния большеберцовой и малоберцовой костей нет. На лопатке при множественных экзостозах типичным является утолщение позвоночного края с неправильными бугристыми наростами. Такие же костные массы наслаиваются и на гребнях подвздошных костей. Головка малоберцовой кости обычно типично вздута в виде канделябра и по своей форме и строению сравнивается с кочаном цветной капусты. В верхней части плеча никогда не приходится видеть экзостозы на узких ножках, как это характерно для проксимального конца большеберцовой кости, — на наружной поверхности плеча экзостозы имеют вид плоских и длинных «табулярных» наростов. Эпифизарные светлые линии на снимках сравнительно мало изменены, обычно они только несколько извиты и неровны. В некоторых случаях эпифизарные диски располагаются наискось по отношению к длиннику кости. В этих случаях имеется дугообразное искривление диафиза. Суставы изменений не представляют, или же экзостозы чисто механически препятствуют полной подвижности. Практически важно, что наружный хрящевой покров экзостоза на рентгенограмме, само собой разумеется, не виден. Поэтому истинные размеры нароста всегда больше, чем рентгенологические, особенно у детей, где хрящевая шляпка может иметь толщину до б—8 мм. Слизистая сумка, образующаяся на месте скольжения мягких тканей над экзостозом (exostosis bursata) и представляющая практический интерес для хирурга, рентгенологически не определяется ни прямо, ни косвенно, за исключением тех случаев, когда имеется обызвествление содержимого сумки. Нам приходилось наблюдать и такие случаи врожденного застарелого заболевания, которые впервые становятся объектом клинического интереса только в результате гнойного бурсита, т. е. вторичного инфекционного процесса в необычной сумочке над экзостозом, и стало быть из-за этого осложнения ставится диагноз основного заболевания. Интересно, что в очень редких случаях наблюдаются переломы ножки экзостоза. Дифференциальная рентгенодиагностика множественных хрящевых экзостозов не существует — слишком характерна рентгенологическая картина этого системного заболевания. Трудности возникают лишь при изолированных асимметричных соли-тарных экзостозах, которые морфологически неотличимы от некоторых форм остеом, остеохондром и хондром. Здесь очень важен не только индивидуальный, но и семейный анамнез. Об отличительном распознавании между экзостозами и доброкачественными опухолями, а также о злокачественном переходе экзостоза речь будет в главе об опухолях. Здесь скажем лишь, что озлокачествление имеет место при множественных хрящевых экзостозах лишь сравнительно редко — скорее всего злокачественность возникает на почве одиночных хрящевых экзостозов. Наиболее часто происходит злокачественный переход при экзостозах бедра, лопатки, таза, позвонков. Гистологически это переход в остеогенную саркому, в хондросаркому, хондромиксосаркому, но и в веретеноклеточную саркому, т. е. злокачественную опухоль самой различной морфологической структуры.
10. МНОЖЕСТВЕННЫЙ ХОНДРОМАТОЗ КОСТЕЙ (БОЛЕЗНЬ ОЛЬЕ)
Множественный хондроматоз костей, также называемый односторонним хондроматозом или дисхондроплазией костей, чаще всего обозначается как «болезнь Олье» по имени известного лионского хирурга Олье (Oilier), открывшего эту нозологическую форму благодаря применению рентгеновых лучей еще в 1899 г. В настоящее время, в век широкого охвата населения рентгенологическим исследованием, болезнь Олье уже никак не может считаться такой редкостью, как это раньше предполагали. Как и все „редкие” болезни, так и множественный хондроматоз костей, при условии достаточного знакомства с ним, несомненно, нередко вылавливается среди других, лучше известных широкому кругу врачей заболеваний опорно-двигательного аппарата. Двадцать лет назад в мировой литературе было описано всего 30 случаев болезни Олье, а в настоящее время количество наших собственных наблюдений значительно, почти в 2 раза, превышает это число. Нет редких болезней, есть редкие диагнозы — это наше суждение и здесь в какой-то мере оправдывается. Множественный хондроматоз скелета является врожденной болезнью, но приобретает клинический интерес в периоде роста, главным образом в детском, реже в юношеском возрасте. Точное начало заболевания в отдельных случаях обычно остается неизвестным, неустанавливаемым, если не было систематического динамического рентгенологического исследования, но это начало относится к очень раннему детству или к внутриутробному периоду. Раньше считалось, что эта болезнь у взрослых людей не встречается. Но, как и ряд других современных авторов, так и мы имеем единичные, действительно редкие наблюдения над болезнью Олье и вне рамок пубертатного возраста, в третьем и даже четвертом десятилетии жизни. Ни разу никто не видел при множественном хондроматозе скелета семейного поражения, это всегда спорадические случаи заболевания скелета. Девочки заболевают по крайней мере в 2 раза чаще, чем мальчики. Сущность множественного хондроматоза костей заключается в каком-то врожденном нарушении нормального процесса роста и развития костного вещества во многих определенных участках зон энхондрального окостенения. Более конкретно можно сказать, что если первая фаза всего процесса окостенения, т. е. пролиферация энхондральных хрящей, и происходит при этой болезни, вероятно, вполне нормально, то именно дальнейшие фазы обызвествления и окостенения развивающегося хряща во время роста кости в длину не наступают, и поэтому преимущественно в мета-дна-физах длинных трубчатых костей остаются не завершившие свой естественный цикл развития необызвествленные и неокостеневшие хрящевые массы. Когда трубчатая кость должна расти в прямом смысле этого слова, т. е. удлиняться, в мета-диафизе остаются обширные хрящевые включения. Микроскопически, по нашим собственным исследованиям, определяются гиалиновые хрящевые островки в различном состоянии развития — в периферических участках хрящевые элементы энергично пролиферируют, а в центральных отделах имеются довольно умеренные дегенеративные явления в хрящевой ткани. Хрящевые массы внутри той или иной кости могут при множественном хондроматозе в дальнейшем разрастаться и в стороны, наподобие опухолей, но ничего общего с истинным бластоматозным процессом болезнь Олье не имеет. Таким образом, болезнь Олье выражается в том, что костная ткань замещена гиалиновым хрящом более или менее правильного строения. Это происходит во многих костях, притом в типичных местах — в метафизарных концах трубчатых костей и в периферических участках плоских костей. Наибольшим изменениям подвергается скелет нижних конечностей, в первую очередь — оба метафиза большеберцовой кости, затем и бедра с преобладанием изменений в его дистальном конце. В гораздо меньшей степени поражаются проксимальный метафиз плеча и дистальные концы костей предплечья, а также оба метафиза малоберцовой кости. И здесь, как и при множественных хрящевых экзостозах, область близ локтя максимально щадится, а близ колена — максимально поражается. В малых трубчатых костях — фалангах пястных и плюсневых костях — изменены и средние трети диафизов. Резкий патологический процесс захватывает тазовые кости, меньше вовлекаются лопатки и ребра. В тяжелых случаях затрагиваются и короткие губчатые кости, как, например, пяточная, таранная и пр. Неизмененными всегда остаются череп, позвоночник и ключицы. Односторонность поражений, которая раньше считалась одним из наиболее важных признаков болезни Олье и даже упоминалась в названии, в настоящее время, в результате более глубокого изучения казуистического материала и накопления большого личного опыта, нами больше не расценивается как обязательный симптом костного хондроматоза, и мы называем болезнью Олье и односторонние, и двусторонние дисхондроплазии. Сейчас известно, что процесс может быть односторонним, т. е. поражать одноименные верхнюю и нижнюю конечности или только одну нижнюю конечность, или же более редко процесс локализуется в обеих нижних конечностях. Наиболее часто встречается двустороннее поражение с более резко выраженным захватом одной стороны, без предпочтения правой или левой половины тела. Бентцон (Bentzon) обратил внимание на тот факт, что хрящевые скопления в костях по локализации и по форме повсеместно совпадают с ходом внутрикостных крупных артериальных стволов и их разветвлений. Во всех костях хрящевые островки, цилиндры и конусы замещают кость, действительно копируя архитектуру внутри-костного артериального дерева. Добившись путем экспериментального повреждения нервных волокон сосудов у кролика превращения костной ткани в хрящевую, приближающуюся по своей рентгенологической и гистологической картине к болезни Олье, Бентцон усматривает сущность дисхондроплазии в каком-то невротрофическом первичном заболевании симпатической нервной системы, к сожалению, более конкретно не разгаданном. Клиническая картина болезни Олье сводится к деформации, развивающейся у ребенка, в остальном вполне нормального, болезненно протекающей и медленно прог рессирующей. Пораженная конечность укорачивается и утолщается, меняется походка, наступает похрамывание. Нередко на первый план выступает варусная или вальгусная деформация колена, реже проксимального конца бедра или стопы со вторичной асимметрией таза и сколиозом позвоночника. Мышечная атрофия отсутствует. Суставы как таковые остаются нетронутыми. Точный диагноз возможен только при помощи рентгенологического исследования. Рентепологическая картина в типичных случаях болезни Олье в высшей степени характерна.
{kind=link}
Рис. 298. Множественный хондроматоз костей (болезнь Олье) у 20-летнего мужчины.
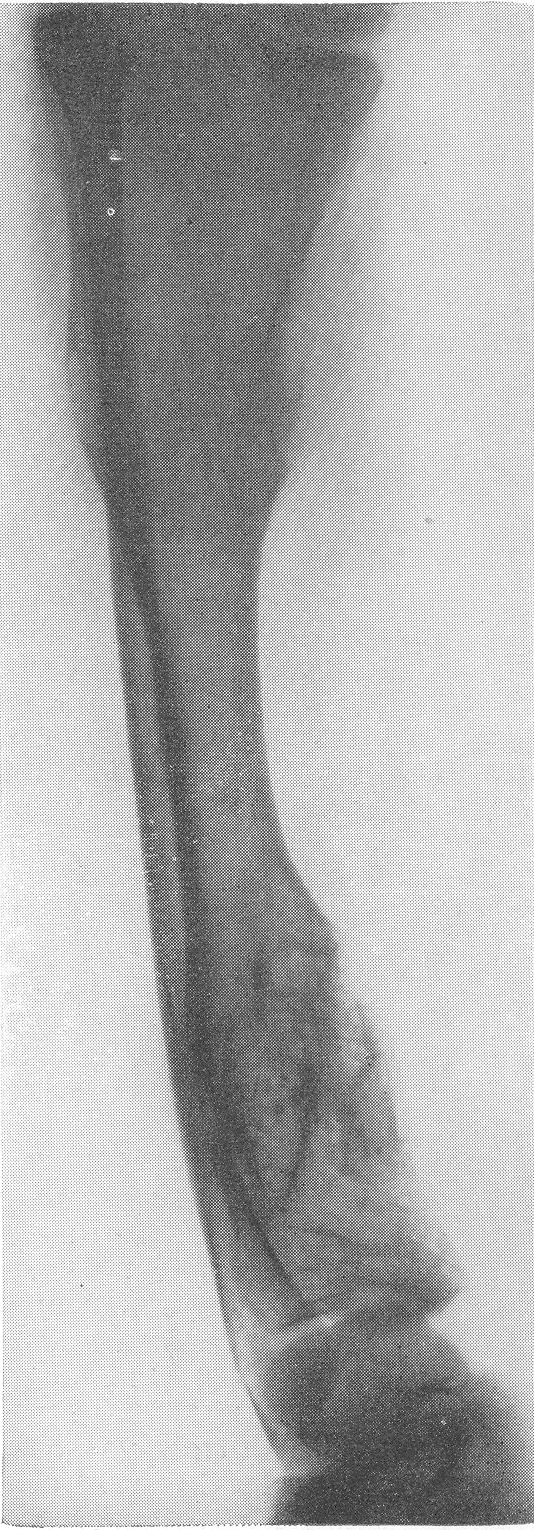{kind=link}
Рентгенограмма голени. Хрящевая перестройка, дугообразная деформация большеберцовой кости, укорочение кости. И на рентгенограммах, соответственно своему анатомическому субстрату, пораженные длинные трубчатые кости умеренно, а иногда и значительно укорочены. В резко выраженных случаях большая трубчатая кость может получить сходство — из-за укорочения диафиза и вздутых эпиметафизарных концов — с гимнастической гирей. Особенно сильно отстают в росте и, стало быть, остаются короткими кости у тех больных, у которых патологический процесс начался очень рано (рис. 261, 3, 297—303).
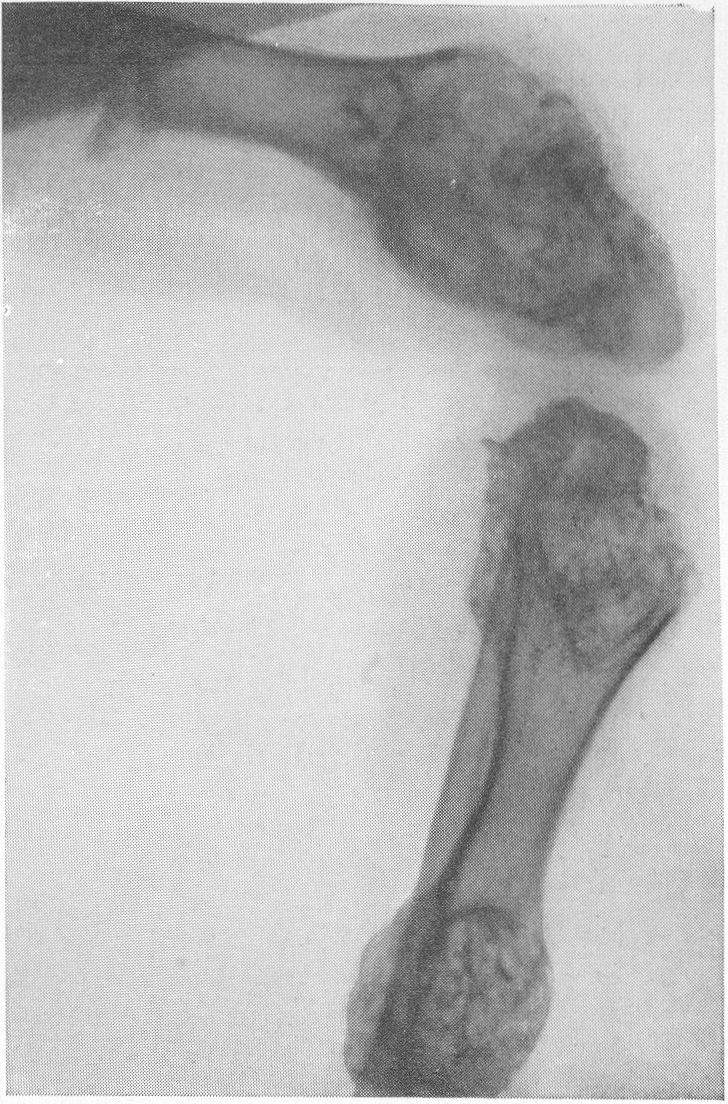{kind=link}
Средние участки диафизов не изменены вовсе или только слегка утолщены. Метафизы же равномерно, или скорее слегка эксцентрически колбообразно, булавовидно расширены, изнутри вздуты. Почти всегда заметно более или менее резкое дугообразное искривление метафизарного конца в сторону от продольной оси кости. Наружные концы кости всегда сохраняют нормальный гладкий вид и четкость. Периост в патологическом процессе участия не принимает. Наиболее характерны структурные изменения, всегда богаче выраженные в большеберцовой кости. Нормальный рисунок метафиза замещен просветлением, продольно расположенным, заостренным по направлению к центру кости и расширяющимся у росткового диска. Это просветление не гемогенно, а исчерчено веерообразно расходящимися в сторону эпифизов костными перегородками, то густо, то редко проведенными, то плотными, склерозированными, то тонкими,
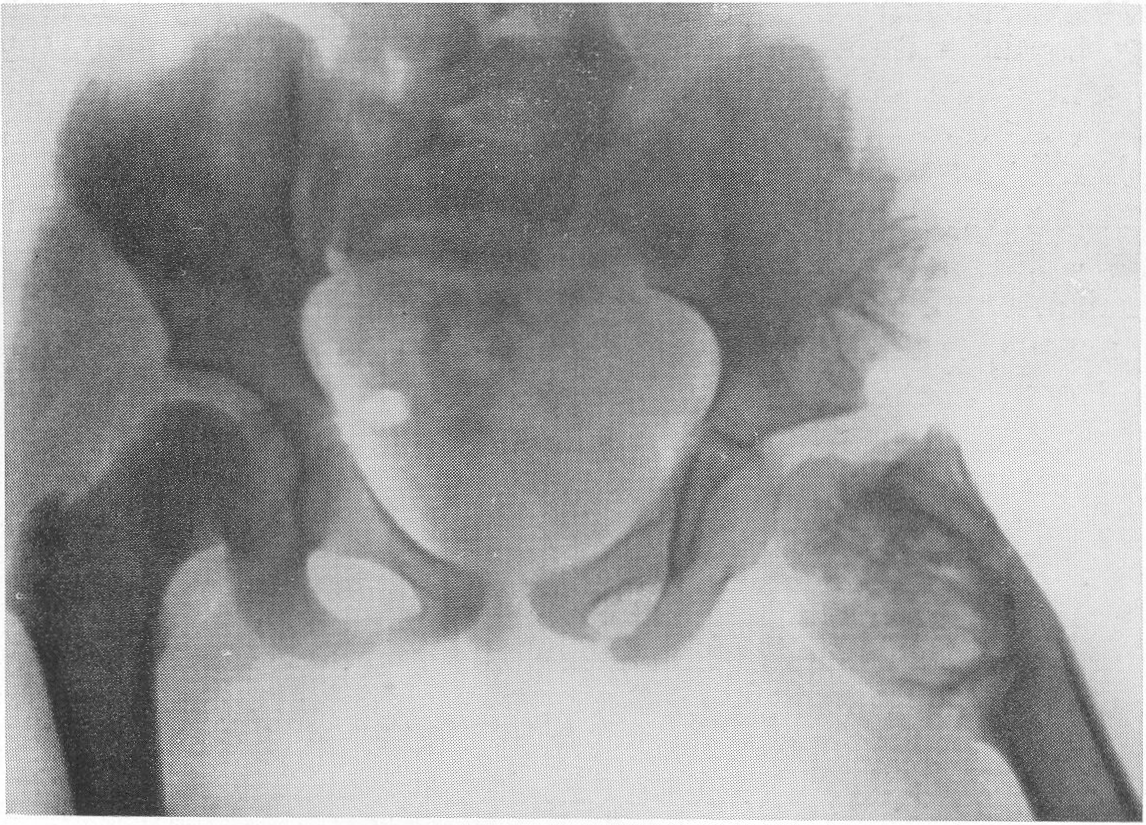{kind=link}
Односторонние изменения (слева) в тазовой кости и в проксимальном метафизе бедренной кости. то длинными, то обрывающимися у начала или конца. Обычно на ряде снимков, произведенных в различных проекциях, можно убелиться в том, что не весь метафиз содержит хрящевую ткань, а просветления лежат по одну сторону от продольной оси кости. Иногда, особенно в дистальном метафизе бедренной кости, хрящевые конические и пирамидальные просветления, разделенные веерообразно идущими тонкими костными „лучами”, спирально изогнуты. Корковый слой над хрящевыми участками большей частью истончен и изнутри приподнят, что и вызывает утолщение метафизов.
{kind=link}
Поражения фаланг и пястных костей двусторонни, но асимметричны, гораздо более выражены справа. На месте прободения корки питающей артерией на тангенциальной рентгенограмме нередко выступает небольшой блюдцеобразный поверхностный дефект с четкими контурами. Эпифизарные линии неровны, змеевидно изогнуты и расположены не перпендикулярно к длинной оси, а слегка под углом. Эпифизы частично сплющены, скошены, что вместе с искривлением метафизов и создает осевые смещения и деформации конечностей. Структура эпифизов ноздревата, кость испещрена множеством округлых и овальных дефектов — хрящевых включений. При незначительной асимметрии ядра окостенения на пораженной или более пораженной стороне имеют меньшие размеры — окостенение, очевидно, замедлено. Большим изменениям, клинически сплошь и рядом мало проявляющимся, подвергаются при болезни Олье фаланги, плюсневые и отчасти также и пястные кости (рис. 301 и 302). Они укорочены, расширены, неуклюжи и содержат множество хрящевых просветлений, то изолированных, то сливающихся друг с другом. Эти дефекты обычно располагаются в центральных участках кости, оставляя корку нетронутой, но иногда они расслаивают кортикальное вещество и несколько вздымают его поверхностные слои. В случае вовлечения и коротких губчатых костей предплюсны, особенно пяточной и таранной кости, в их структуру вкраплены точечные просветления. Из плоских костей наибольшие превращения, притом рентгенологически специфические, претерпевают кости таза. Подвздошные кости лишь в центре сохраняют нормальный структурный рисунок. Поверхностные же участки заняты светлыми овалами и треугольниками, отделенными друг от друга балками, рассыпающимися в виде лучей наподобие остова крыла бабочки.
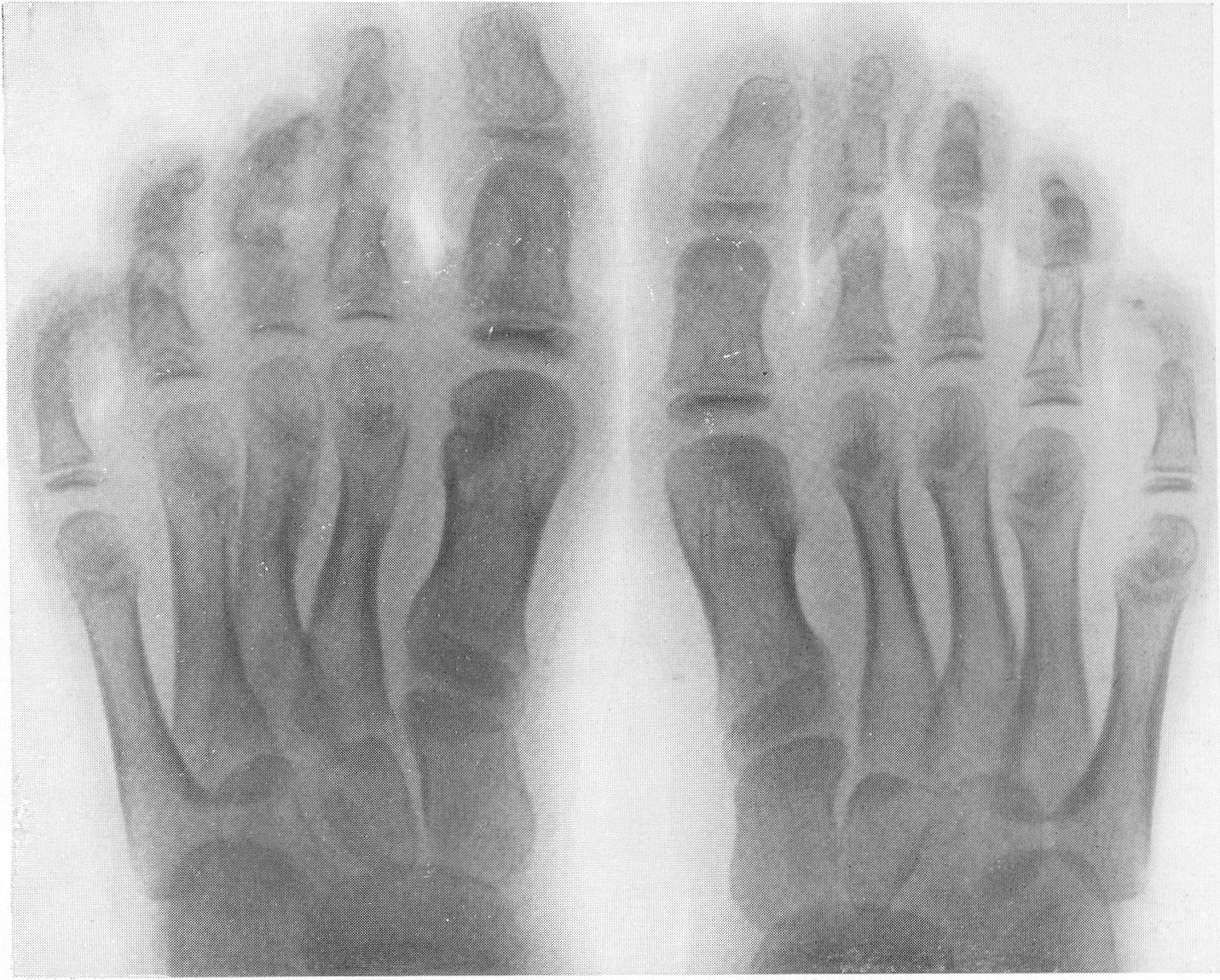{kind=link}
Одностороннее поражение ряда фаланг и плюсневых костей слева у 9-летнего ребенка. То же самое можно наблюдать и на рентгенограмме лопатки, правда, не в столь чистом виде. Дефекты в окружности запирательного отверстия могут располагаться так густо, что теряется дифференциация между корковой и губчатой структурой. Дно вертлужной впадины показывает своеобразный ноздреватый рисунок, который может быть заметен также и в области шейки лопатки. Ребра — не все, а только единичные с обеих сторон — своими передними концами расширены в виде раструбов, иногда расщеплены. Межреберья местами расширены, местами сужены, что, впрочем, нисколько не меняет общей правильной конфигурации грудной клетки. Патологических переломов при болезни Олье, как это и следует ожидать на основании теоретических предпосылок, на местах хрящевых включений не бывает. Общая принципиальная рентгенологическая симптоматология при множественном хондроматозе костей весьма однообразна и характерна, и поэтому дифференциальная диагностика в типичных случаях болезни Олье после рентгенографии, можно сказать, не существует. Однако каждый отдельный случай заболевания в такой степени крайне индивидуален и неповторяем в своих частных особенностях и в сочетаниях болезненных признаков, что практически все же заставляет нас проводить отличительное распознавание. Такие клинические диагнозы, как атипичная хондродистрофия, частичная гемигипертрофия или гемиатрофия костей, рахитические деформации или обычные genua valga и vara сразу же отвергаются с рентгенологической стороны. Элементарно легко отбросить на основании снимков предположение о болезни Реклингаузена, с которой чаще всего в практической жизни смешивается болезнь Олье при недостаточном знакомстве с нею. Главные затруднения, иногда непреодолимые, возникают лишь в отличительном распознавании между слабо выраженной „абортивной формой” болезни Олье (рис. 303) и истинными опухолями — хондромами в случае изолированного поражения скелета кисти или стопы. Здесь первым делом должен быть устранен тот источник ошибок, который заключается в рентгенографии одной только области скелета, а не всего скелета. Хондромы обычно разбросаны крайне асимметрично, они достигают значительных размеров, шаровидны, ведут к девиациям костей, растут эксцентрично, а главное, щадят при множественном поражении малых трубчатых костей большие трубчатые кости. Имея перед собой один только снимок кисти или стопы с небольшими центрально сидящими хрящевыми дефектами, рентгенолог, следовательно, вправе отказаться от дифферециальной диагностики между болезнью Олье и бластомой. Такова же постановка вопроса при исключении множественного кистовидного туберкулезного остита Юнглинга и саркоидоза. В нашей консультативной практике мы также видели случай смешения хрящевого островка в области шейки бедра при болезни Олье с изолированным туберкулезным очагом, случай поражения дистального конца бедренной кости с клиновидным секвестром при остеомиелите, принятый до нас ошибочно за хондроматоз, а также случай болезни Олье, ложно распознанный и энергично леченный рентгеновыми лучами как костная гемангиома.
{kind=link}
Рис.303. Множественный хондроматоз костей (болезнь Олье).
Небольшой клиновидный участок поражения в медиальной части проксимального мета-диафиза правой плечевой кости может создать дифференциально-диагностические трудности. Вопрос решается при рентгенографии периферических участков конечности, всегда больше пораженных. Вопреки мнению Фербенка (Fairbank), мы считаем болезнь Олье совершенно не связанной со множественными хрящевыми экзостозами. Это вовсе не варианты или разновидности одного и того же заболевания, которое предлагают объединить общим названием множественная метадиафизарная дисплазия. Действительно общим следует считать только множественные врожденные нарушения процесса костно-хрящевого развития скелета. Изменения эти при множественных хрящевых экзостозах обращены, если можно так выразиться, наружу, а при болезни Олье — внутрь кости. Вопреки литературным указаниям на сочетание обеих болезней, мы никогда не наблюдали комбинированных форм или перехода одной формы в другую. Поэтому мы и считаем, что во всяком случае с клинических и рентгенологических позиций обе болезни резко отличаются друг от друга, могут и должны быть в порядке дифференциальной диагностики противопоставлены. Болезнь Олье также никогда не наблюдается в семьях, страдающих множественными хрящевыми экзостозами.
Необходимо еще иметь в виду, что костная болезнь Олье в довольно редких случаях сочетается с поражениями других систем. Мы б раз наблюдали типичные комбинации множественного хондроматоза костей с кавернозными гемангиомами и флеболитами в этих кровяных пазухах. Это так называемый синдром Мафучи (Maffucci), описанный этим автором в 1881 г. Можно не сомневаться в том, что в основе синдрома Мафучи лежат глубокие изменения нервной системы, именно вегетативной нервной системы. Мы также наблюдали сочетание болезни Олье с тератомой яичника у маленьких девочек со значительными эндокринными нарушениями — преждевременным, очень ранним (в возрасте 21/2 лет!) включением правильных менструаций и половым созреванием и соответствующим ускорением окостенения, причем после оперативного удаления опухоли яичника общее развитие вернулось к норме. Общий прогноз при множественном хондроматозе скелета благоприятен. Что касается возможности озлокачествления при болезни Олье, то в этом в настоящее время больше сомнений нет. Впервые в отечественной литературе малигнизация описана в 1956 г. Н. М. Енгалычевой. Мы сами наблюдали один чрезвычайно убедительный случай (опубликованный Т. И. Глинер и Н. И. Сеферовой) у 25-летней женщины, у которой типичная болезнь Олье стала клинически проявляться в возрасте 3 лет, затем в 5-летнем возрасте была нами точно распознана рентгенологически. Крупный злокачественный опухолевый узел развился у больной быстро после беременности и лактации в области диафиза плечевой кости, был оперативно удален и при гистологическом контроле оказался хондросаркомой с большим центральным некротическим распадом и ослизнением (рис. 304). Не подлежит сомнению, что при синдроме Мафучи малигнизация наступает значительно чаще, чем при обычной неосложненной болезни Олье.
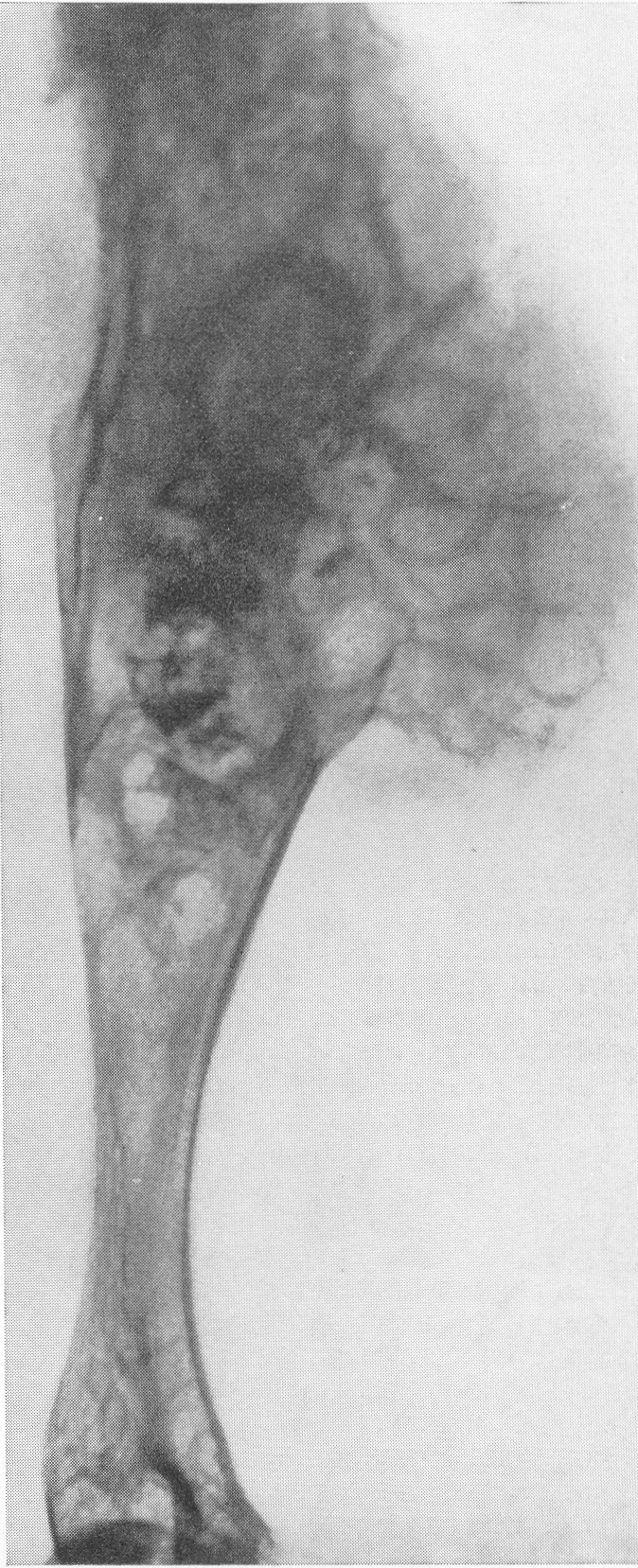{kind=link}
Рис. 304. Множественный хондроматоз костей (болезнь Олье). Озлокачествление одного из костно-хрящевых узлов в области диафиза плечевой кости у 25-летней женщины, у которой диагноз основного заболевания был поставлен 20 лет назад. Гистологическое подтверждение диагноза (остеохондромиксосаркома). Благополучие через 3 года после удаления малигнизировавшегося участка.
Рационального лечения болезни Олье не существует, влиять активно на течение патологического процесса мы не умеем. Имеются указания на то, что с течением времени, обычно ближе к пубертатному возрасту, хрящевая ткань при болезни Олье заменяется костной и хрящевые просветления на сериях рентгенограмм уменьшаются. Вполне вероятно, что именно благодаря подобному процессу самоизлечения болезнь Олье так редко наблюдается у взрослых. Функциональный прогноз у маленьких детей омрачается пусть медленно, но все же прогрессирующей деформацией. Лечение может быть только симптоматическим, ортопедическим. От оперативного исправления деформаций в школьном возрасте мы удовлетворительных стойких результатов не видели и поэтому убежденно считаем, что хирургическое вмешательство в лучшем случае показано лишь у взрослых больных, для исправления уже сформировавшихся, а не динамических деформаций.
11. ВРОЖДЕННАЯ МРАМОРНАЯ БОЛЕЗНЬ
Врожденная мраморная болезнь описана впервые в 1904 г. гамбургским рентгенологом Альберс-Шенбергом (Albers-Schonberg) и в литературе теперь широко обозначается именем этого исследователя, одного из пионеров мировой рентгенологии, — болезнь Альберс-Шенберга. Другими названиями этой болезни являются остеопетроз (окаменелость костей), генерализованный или системный остеопетроз, врожденный генерализованный или системный остеосклероз. Некоторый смысл имеет противопоставление врожденной генуинной мраморной болезни симптоматической мраморной болезни, о чем речь будет подробно впереди. Врожденная мраморная болезнь относится к разряду довольно редких нозологических единиц. Это удивительное системное поражение скелета, так живо действующее на воображение врачей и поэтому охотно описываемое в виде казуистических сообщений, в настоящее время насчитывает в мировой литературе больше 300 случаев. Болезнь Альберс-Шенберга имеет в подавляющем большинстве случаев явно семейное выражение, передается в той или иной степени из поколения в поколение, однако сплошь и рядом попадается и в виде спорадического поражения человека из здоровой семьи. Врожденная мраморная болезнь наблюдается в любом возрасте. Единичные случаи распознаны рентгенологически даже у плода, т. е. еще в утробе матери. Тяжело протекающие случаи сказываются в первое время после появления новорожденного на свет, более легкие — в детстве, а самые доброкачественно текущие — в зрелом и даже старческом возрасте, например в восьмом десятилетии жизни. Течение и предсказание в значительной степени связаны с возрастом, чем раньше проявляется эта врожденная болезнь, тем хуже предсказание, а у взрослых людей общий прогноз расценивается как весьма благоприятный. Можно, таким образом, говорить о благоприятных и о неблагоприятных формах врожденной мраморной болезни. Этиология врожденной мраморной болезни все еще неизвестна. Состояние нервной системы еще никем надлежащим образом не подвергнуто всестороннему изучению. Эндокринные железы при гистологическом исследовании всегда оказываются без существенных патологических сдвигов. Несомненно, что эта болезнь означает какие-то глубокие изменения минерального, в частности кальциевого и фосфорного обмена. Большинство современных авторов смотрит на врожденную мраморную болезнь как на своеобразную недостаточность мезенхимы, как на способность мезенхимы связывать чрезмерное количество качественно измененных солей. Имеются указания на очень глубокие изменения минерального состава костного вещества, на нарушение нормального баланса различных видов кальция в костной субстанции. В основе заболевания лежит неправильный процесс окостенения, выражающийся в том, что вырабатывается увеличенное количество компактного костного вещества. Повышенное производство костных элементов происходит главным образом на месте эндостального, а также энхондрального роста кости. На вскрытии кость имеет равномерную бледно-серую окраску и напоминает однородный мрамор. Теряется нормальное разграничение между компактным и губчатым костным веществом. Под микроскопом определяется неправильная, беспорядочная, какая-то бессистемная гистологическая архитектоника костного вещества. Количество остеобластов нормально или увеличено, количество остеокластов же нормально или понижено, вплоть до их полного отсутствия. Характерна густая сеть качественно недоразвитого и неполноценного хондро-остеоидного вещества. Надкостница, как правило, нормальна. Имеются, впрочем, данные о фиброзной метаплазии в некоторых случаях заболевания. Важно, что резко понижено кровоснабжение кости, васкуляризация сильно ограниченна, нарушена в значительной степени и иннервация костной системы. Корковое вещество в 2 или 3 раза толще, чем в норме, и это утолщение происходит почти исключительно в сторону костномозгового канала. Последний, следовательно, резко суживается или полностью зарастает, и тогда весь костный ствол становится однородным. Все пазухи и щели между трабекулами губчатого вещества заполняются новыми балочками, старые трабекулы утолщаются благодаря наслоению новых пластов костного вещества, и губчатое костное вещество превращается почти повсеместно в компактное. На местах энхондрального окостенения хрящевое вещество пропитывается известью в чрезмерной степени, зона предварительного обывествления резко расширяется, нормальный процесс рассасывания извести в субхондральном костном отделе выпадает. Эпифизарная линия имеет неровный извилистый вид. Вытеснение костного мозга, а также его замещение фиброзной соединительной тканью ведут к анемии вторичного, гипохромного типа. Надо думать, что этот же процесс механического вытеснения костного мозга служит причиной вторичной глубокой перестройки других звеньев кровотворного и ретикуло-эндотелиального аппарата. Создаются дополнительные центры кровотворения, развивается спленомегалия, увеличиваются порой значительно печень, а в меньшей степени и лимфатические узлы. Остающийся костный мозг функционально активен. В периферической крови число лейкоцитов повышено у детей, понижено у взрослых. Могут попадаться незрелые элементы, например, в первую очередь нормобласты. В тяжелых случаях, главным образом у детей, изменения кровотворного аппарата развиваются подчас внезапно, прогрессивно и бурно, этим самым становясь непосредственной причиной смерти, в других же случаях, преимущественно у взрослых, анемия держится годами без того, чтобы больные от этого испытывали субъективные неприятности. Кальций и фосфор крови обычно нормальны или колеблются в небольших пределах в ту или иную сторону вокруг нормы. Обмен веществ, определенный обычными клиническими методами исследования, существенных сдвигов не представляет. Бывает кровоточивость. Общее развитие больных несколько страдает. Больные низкого роста, грацильны, худы, поздно начинают ходить. У детей часто бывает гидроцефалия. Если болезнь проявляется в детском возрасте, эпифизарные ядра окостенения будто запаздывают, и синостоз наступает несколько позже, чем у нормальных сверстников. Из-за остеосклероза различные костные каналы, полости и отверстия суживаются. Давление на зрительные нервы может послужить причиной ослабления зрения или полной слепоты, наблюдается нистагм, паралич глазных мышц. Встречаются и поражения лабиринта и т. д. Зубы появляются очень поздно и очень плохо развиты, легко поражаются кариесом, выпадают (рис. 314), их инфекция в некоторых случаях ведет к остеомиелиту и к некрозу нижней челюсти — смертельному осложнению. Описаны случаи комбинации мраморной болезни костей с обызвествлением сосудов и опорной соединительной ткани, а также с так называемыми метастазами кальция во внутренние органы. Точное распознавание врожденной мраморной болезни на основании одних только клинических признаков почти невозможно, во всяком случае никогда не надежно. У детей на первый план в клинической картине выступают вовсе не основные костные проявления болезни, а только такие вторичные процессы и осложнения ее, как гидроцефалия, нарушения со стороны глаз— атрофия зрительных нервов, стоматологические симптомы. Они-то и служат важными показаниями к рентгенологическому исследованию. И только при первом в жизни рентгенологическом вмешательстве — как мы видели, еще до появления на свет или же в любом возрасте в длинной человеческой жизни — открывается обычно неожиданно основное скелетное заболевание. В очень доброкачественно протекающих случаях у взрослых людей мраморная болезнь вообще может протекать без серьезных клинических проявлений и без соответствующей анамнестической поддержки. Если не было основания для рентгенологического контроля состояния того или иного органа или непосредственно скелета, то болезнь может остаться вообще при жизни нераспознанной. Она тогда не называется своим настоящим именем и на секционном столе, за редчайшими исключениями. Рентгенологическая картина мраморного скелета в высшей степени оригинальна (рис. 261, И), только благодаря рентгенологическому исследованию мраморная болезнь и стала известна медицинскому миру. Больше и сильнее всего поражаются череп, таз (рис. 305 и 310), из больших трубчатых костей — проксимальные концы бедренных и дистальные концы берцовых костей, ребра, позвонки (рис. 306 и 312). Меньше изменены плечевые кости, фаланги. Внешняя форма и размеры костей не изменены вовсе, или же эпифизарные концы костей несколько утолщены и закруглены. Метафизы также больше изменены, чем диафизы, — они тоже немного неуклюжи, булавовидно утолщены.
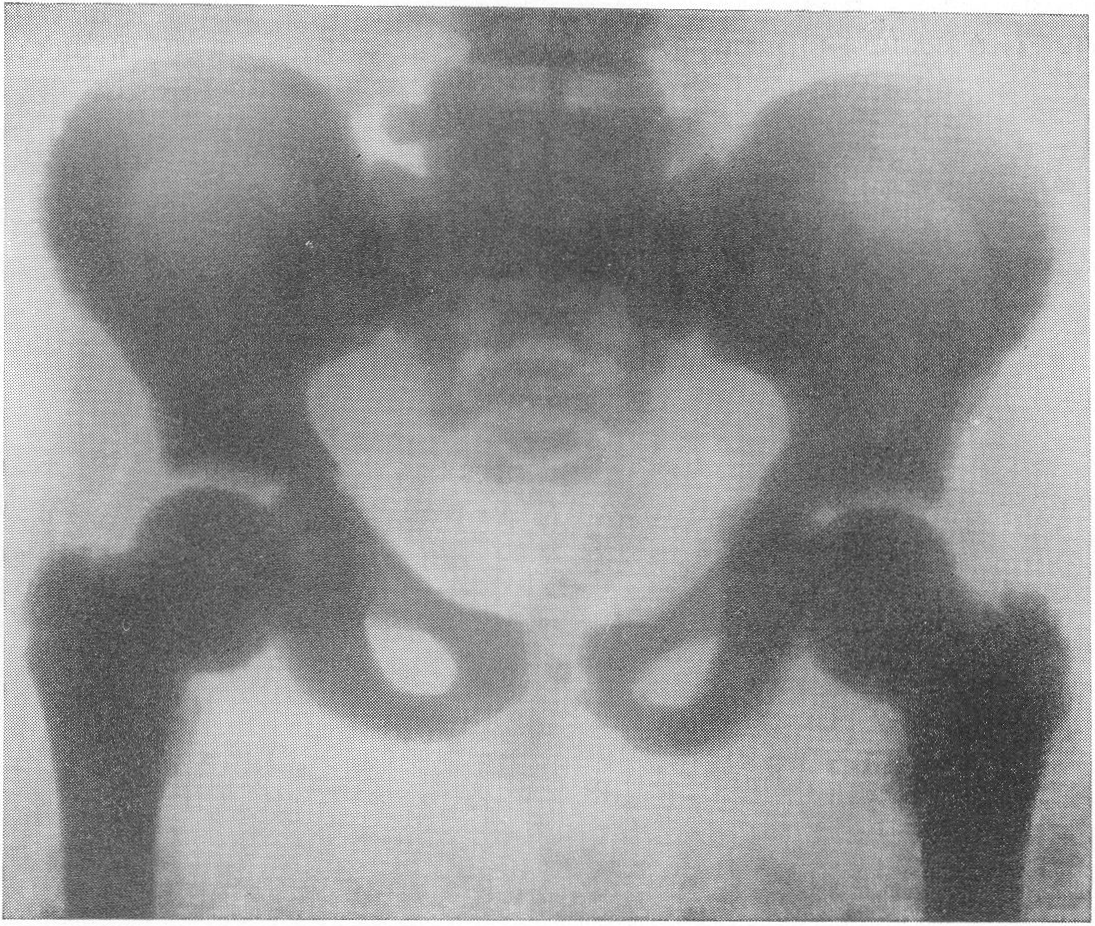{kind=link}
Рис. 305. Врожденная мраморная болезнь у 10-летнего мальчика. В анамнезе — несколько переломов костей, патологический характер которых был просмотрен вследствие неприменения рентгенологического исследования. Нормальная картина крови. Нормальные биохимические показатели. Рентгенограмма таза.
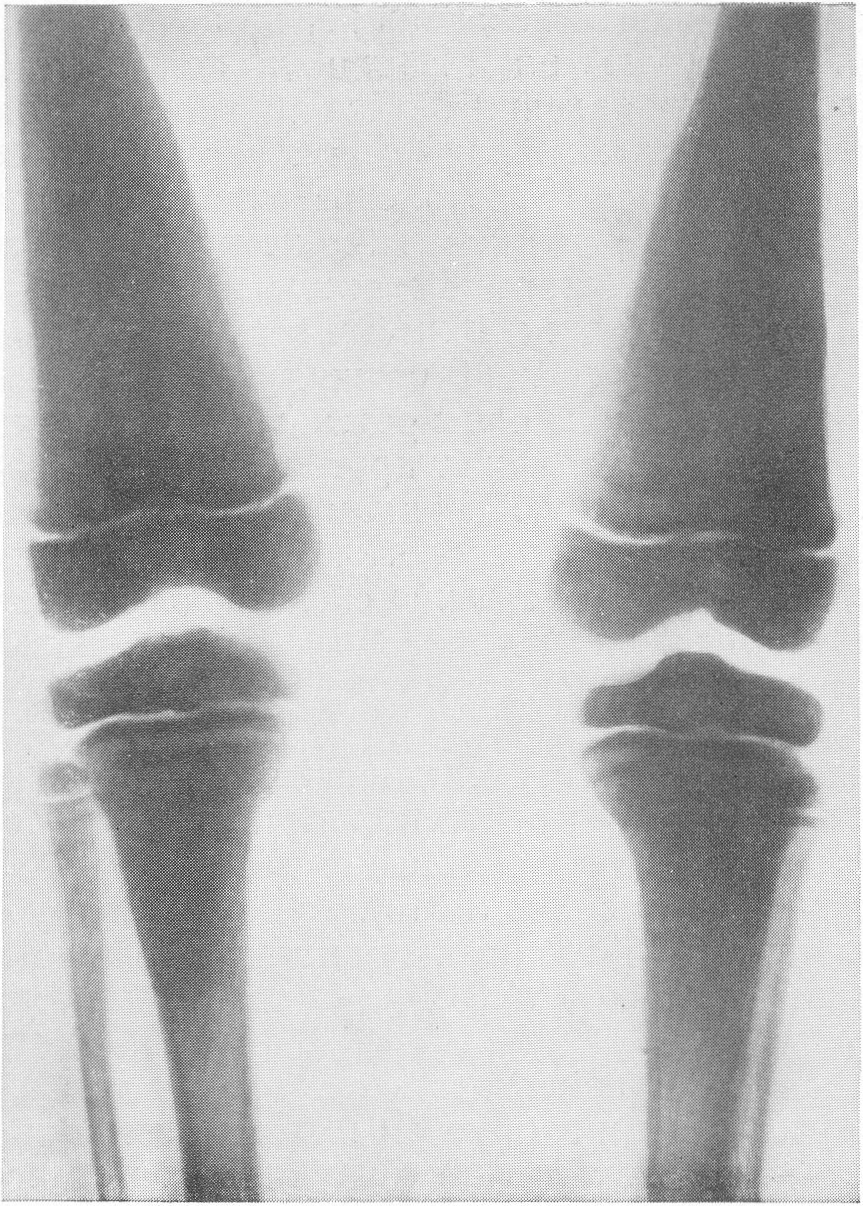{kind=link}
Все кости представляются плотными и непрозрачными для рентгеновых лучей, так что с первого взгляда снимки производят впечатление слишком мягких или недоэкспонированных, бракованных. Благодаря мощным наслоениям компактного костного вещества со стороны эндоста костномозговой канал отсутствует полностью или едва выступает в виде центральной узкой продольной щели (рис. 311). Отсутствует и губчатое строение, эпифизарные концы костей и мелкие кости дают интенсивную вполне гомогенную сплошную тень на негативе, в действительности имеющую сходство с белым мрамором. В некоторых случаях диафизы или метафизы длинных трубчатых костей сохраняют частично нормальное строение. Тогда на рентгенограммах получаются широкие поперечные светлые зоны, резко выступающие на фоне темной плотной мраморной кости. То же самое наблюдается и в плоских и коротких губчатых костях, в особенности костях предплюсны, как в пяточной или таранной костях; центральные части сохраняют нормальный вид или заняты отдельными изолированными островками компактного вещества, а поверхностные части склерозированы в виде широкого пояса, имеющего форму, соответствующую наружной конфигурации кости (рис. 308). Иногда светлых поперечных зон в больших костях и концентрических поясов в коротких несколько, т. е. широкие светлые зоны чередуются с темными бесструктурными — выражение ремиссий и рецидивов, известного нарастания активности костеобразования, сменяющегося ослаблением его. Это характерная для ряда случаев мраморности костей полосатость, стратификация. В очень редких случаях эта полосатость бывает не поперечной, а довольно отчетливо продольной, осевой, при различной ширине светлых и темных полос. В телах позвонков полосатость всегда горизонтальная — темные полосы вдоль площадок, светлая посередине тела (рис. 309—313).
Пневматические полости черепа, особенно лобные, при болезни Альберс-Шенберга целиком заращены такой же плотной бесструктурной костной тканью.
{kind=link}
Гипофизарная ямка мала, уплощена. Processus clinoidei posteriores булавовидно вздуты. Особенно плотны кости основания черепа (см. рис. 310), позвонки и тазовые кости. Одним из наиболее постоянных, хотя и не абсолютно обязательных осложнений мраморной болезни, которое приводит больного к рентгенологу и этим самым дает возможность неожиданно обнаружить и основную болезнь, являются патологические переломы. В происхождении этих переломов нет ничего удивительного — повышенное содержание минеральных солей и увеличенное количество костных элементов совершенно не равноценны, как это могло бы показаться с первого взгляда, усиленной механической стойкости кости. Остеосклероз ведет к уменьшению эластичности и упругости кости и служит причиной ее ломкости. Собственно говоря, вид тяжелой на вес кости при мраморной болезни на секционном столе и особенно в рентгеновском изображении создает ложное внешнее впечатление, в действительности эта „мраморная” кость хрупка скорее, как палочка мела.
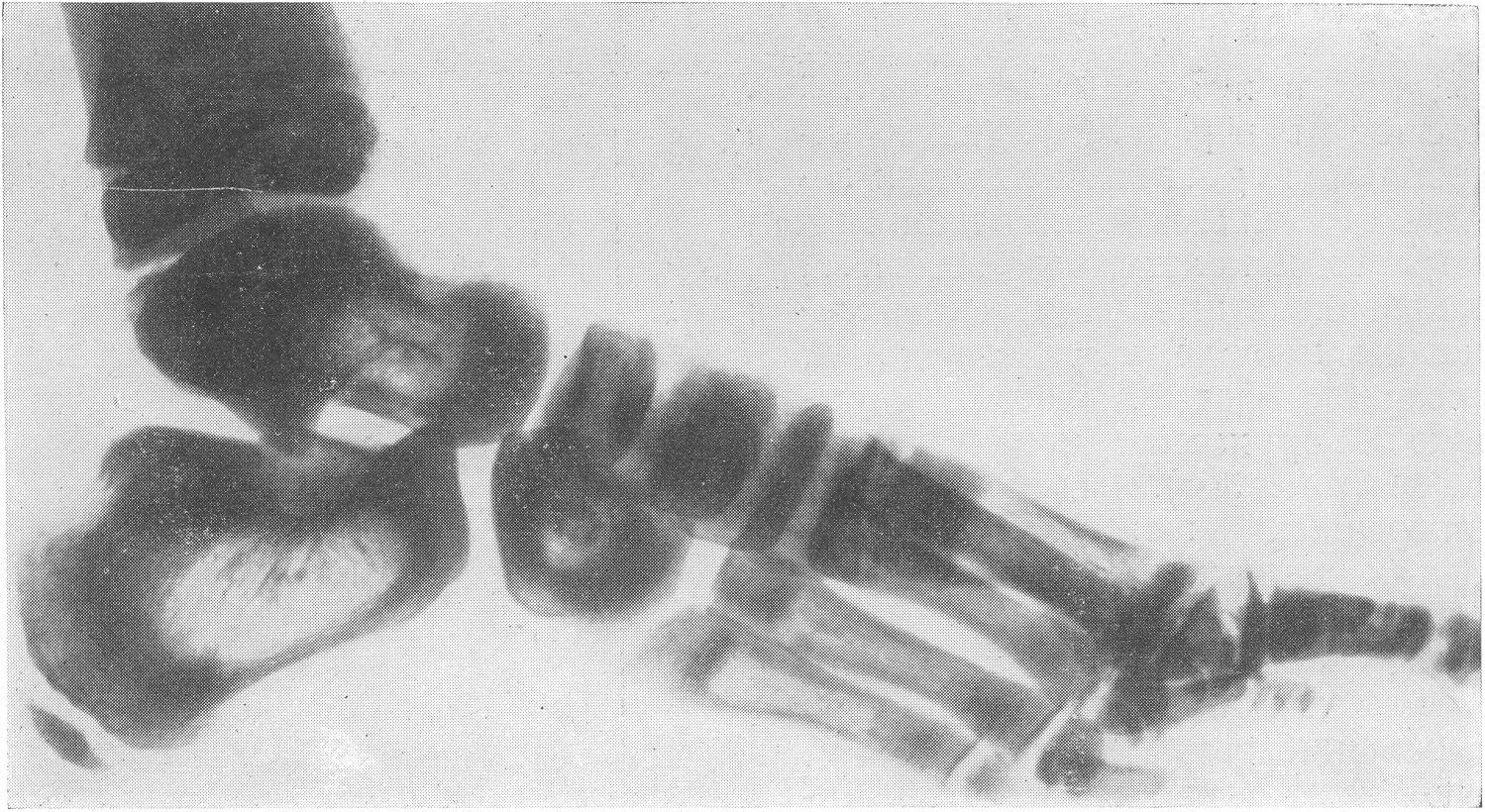{kind=link}
Рис. 308. Тот же больной. Рентгенограмма стопы. Полосатость (стратификация) рисунка костей предплюсны. Центральное затемнение в виде ядра в кубовидной кости.
{kind=link}
Рис. 309. Тот же больной. Задняя (А) и боковая (Б) рентгенограммы грудного отдела позвоночника. Резко выраженная полосатость (стратификация).
Просверлить эту кость дрелью или ее распилить гораздо легче, нежели кость нормальную. В этом отношении крайности — остеосклероз и остеопороз — сходятся. При мраморной болезни чаще всего ломаются кости нижних конечностей, особенно бедра под малым вертелом. Заживление переломов происходит вполне нормально, вероятно, благодаря нормальному состоянию надкостницы. Впрочем, наблюдается и ускорение заживления, но и чаще замедление, и последнее может быть объяснено наблюдаемой подчас фиброзной перестройкой надкостницы. Довольно обычны также деформации, вызванные не только переломами, но и различными неправильностями развития в местах энхондрального роста. Сюда относятся деформации лицевого и мозгового черепа, искривление позвоночника, деформации ребер и грудной клетки, coxa vara и пр. Большие трубчатые кости всегда сохраняют свою нормальную длину, но в некоторых случаях (особенно бедренные кости) могут быть слегка дугообразно искривлены. Точнее было бы сказать, что нормальные кривизны усугубляются до высших пределов.
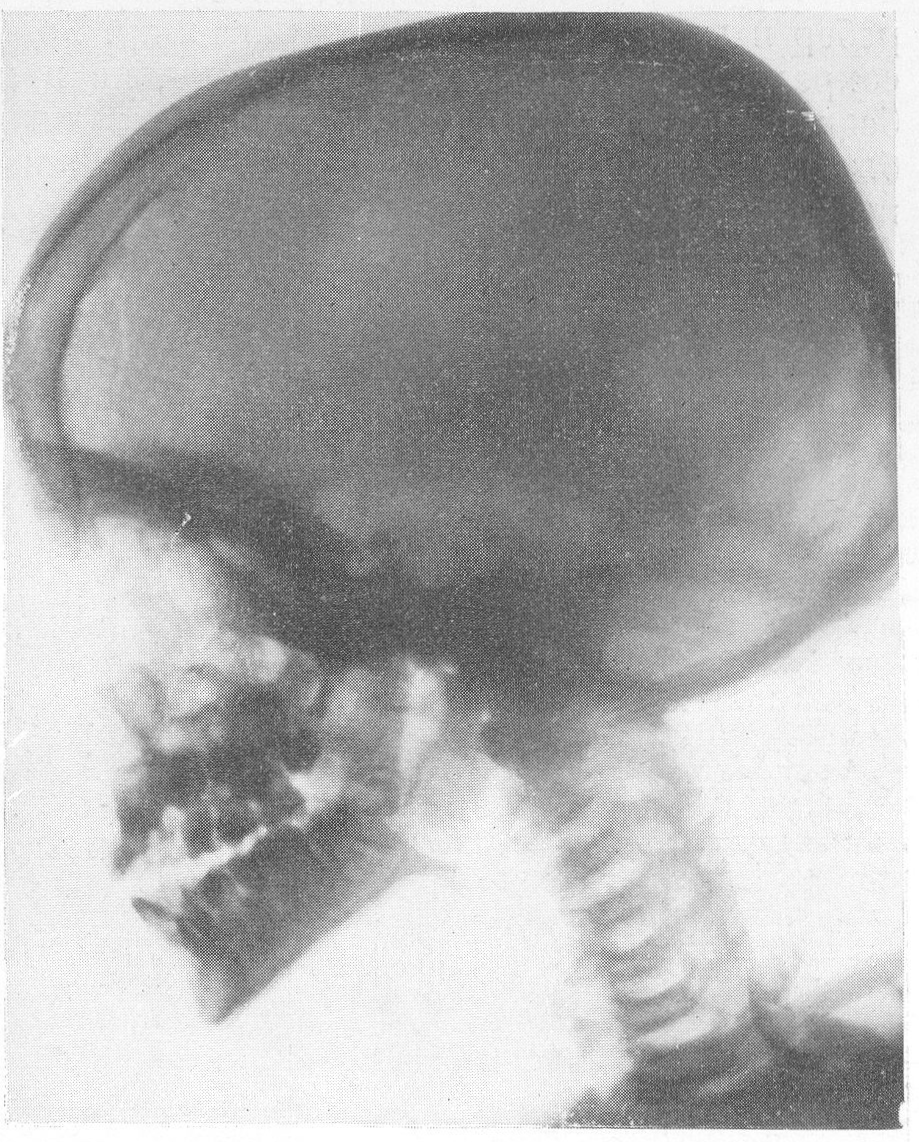{kind=link}
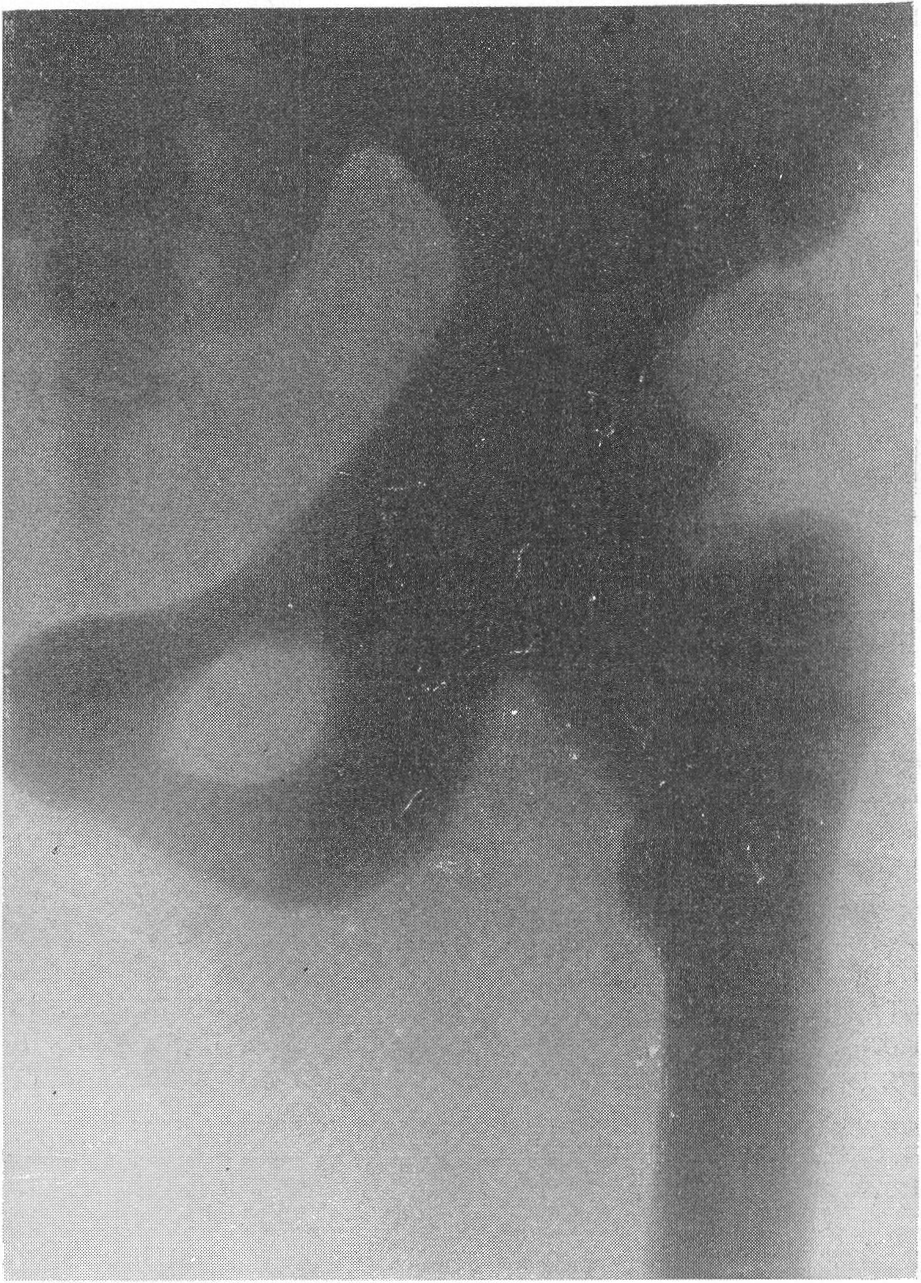{kind=link}
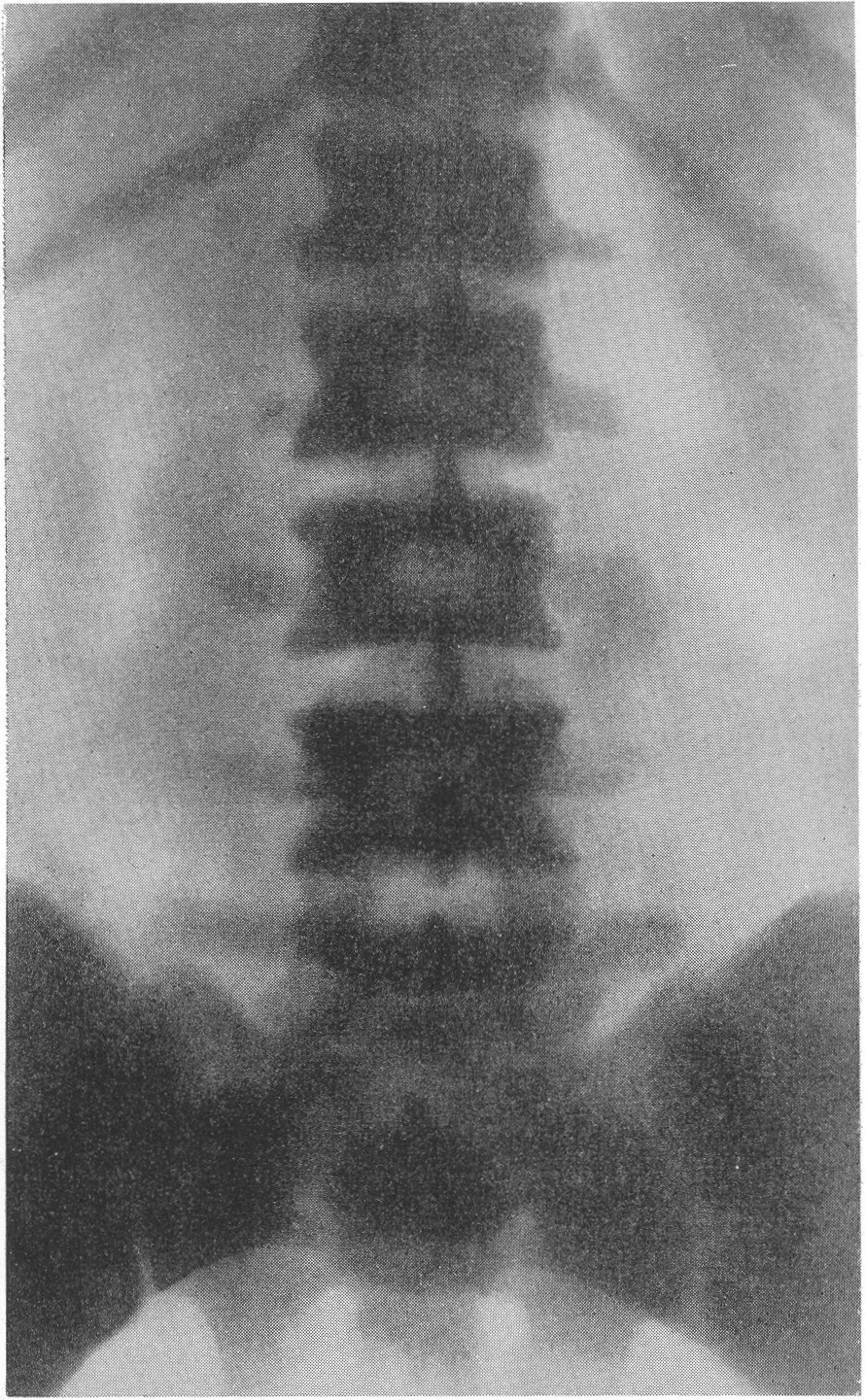{kind=link}
Дифференциальная диагностика врожденной мраморной болезни с ее большим возрастным диапазоном и многообразными клиническими проявлениями была бы крайне трудной, если бы она не опиралась на богатую и характерную рентгенологическую картину. Конечно, как всегда, в общем сочетанном отличительном распознавании немалое значение имеет и довольно четко определившийся в настоящее время клинический фон заболевания — семейный и личный анамнез, перенесенные в прошлом переломы костей, глазные и челюстные проявления, факты из области гематологии, отрицательные биохимические показатели и т. д. Но в свою очередь и сама рентгенология выдвигает сейчас требование перебрать и исключить достаточно длинный ряд различных по природе болезней, приближающихся по тем или иным своим симптомам к врожденной мраморности скелета. Естественно, что речь идет о тех патологических процессах, которые так или иначе ведут к остеосклерозу, особенно распространенному или системному.
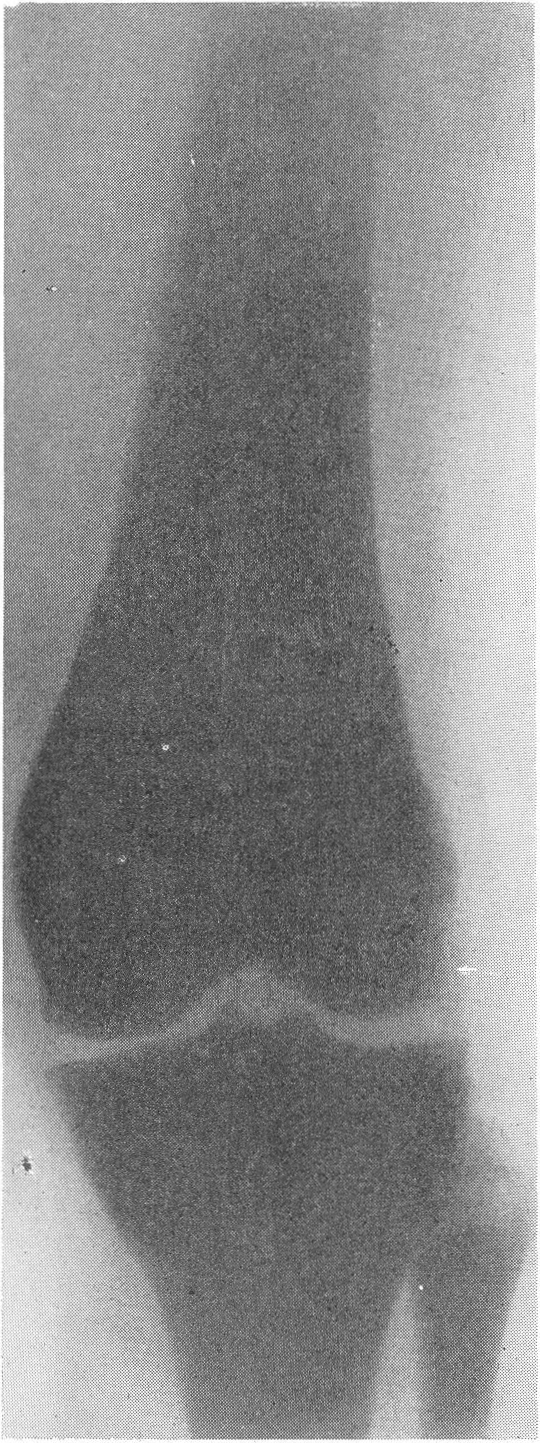{kind=link}
Самым важным в практической дифференциальной рентгенодиагностике врожденной мраморной болезни является исключение обширных остеосклерозов, развивающихся вторично в качестве костной реакции при системных поражениях костного мозга — при миелофиброзе, остеомиелосклерозе. Несомненно, что ряд описанных в литературе случаев мраморности скелета относится именно не к врожденной, здесь описываемой, если можно так условно выразиться, первично-костной мраморной болезни (на самом деле, само собой разумеется, все остеосклерозы по существу вторичны!), а именно к явно вторичному костному процессу — при миелофиброзе. О нем, как и о других ниже упоминаемых патологических формах, подробно говорится в соответствующих разделах. У взрослых больных надо помнить об остеосклерозах при хронической почечной недостаточности, как одной из разновидностей остеонефропатии. Значительные распространенные остеосклерозы, подчас переходящие по интенсивности уплотнения костного вещества в однородную мраморность, могут развиваться в некоторых случаях, в соответствующих возрастных группах, обезображивающей остеодистрофии Педжета, а также при метастатическом карцинозе скелета, обусловленном главным образом первичным раком предстательной железы. Остеосклероз может быть вызван в редчайших случаях особой формой миеломной болезни. То же самое может случиться и в редких случаях лейкоза и лимфогранулематоза. Из эндокринных болезней надо учесть остеосклерозы при гипопаратиреозе во всех его разновидностях.
{kind=link}
Большой теоретический и практический интерес представляют системные остеосклерозы при хроническом действии на скелет таких внешних факторов, как соли тяжелых металлов, некоторые галоиды и т. п. (например, влияние свинца, фосфора, особенно же фтора). Серьезное дифференциально-диагностическое значение в первые дни, недели и месяцы жизни принадлежит той форме врожденной остеонефропатии, которая называется идиопатической гиперкальциемией раннего детского возраста. В детском возрасте надо иметь в виду также врожденный сифилис с его костными особенностями под влиянием сильно действующих лекарственных веществ, равно как и изменения костной системы при гипервитаминозе А и D. Из группы врожденных системных заболеваний могут приобрести некоторое значение подчас мелореостоз, остеопойкилия и множественные диафизарные гиперостозы. Практически актуальны также ограниченные остеосклерозы при заболеваниях кровотворной и ретикуло-эндотелиальной систем, особенно при лимфогранулематозе, которые вызывают местные картины мраморности, например пресловутые „мраморные позвонки”. Наконец, следует подчеркнуть во избежание ошибок при дифференциальной диагностике, что врожденная мраморная болезнь вовсе нередко осложняется остеомиелитом и рахитом, и поэтому обоснованное определение этих последних болезней отнюдь не исключает мраморной болезни, а означает их сосуществование. Лечение врожденной мраморной болезни по существу еще не в нашей власти. Ведь какие-то глубокие анатомо-физиологические сдвиги в целостном организме при течении этой болезни несомненно происходят, иногда болезнь как бы самостоятельно прекращается, обрывается или, наоборот, усиливается, бывают ремиссии и так называемые интермиссии. Очевидно, что действуют какие-то факторы внешней среды. Но мы их, к сожалению, еще конкретно учитывать, а потому и применять не умеем. Мы в настоящее время здесь еще не умеем активно воздействовать на организм с целью восстановить нормальные процессы костеобразования. Казалось бы, что следует постараться мобилизовать избыточные минеральные отложения из костных депо. Но все попытки подобного рода до сих пор успехом не увенчались. Ясно, что при врожденной мраморной болезни надо избегать введения в организм всех тех веществ, которые способны стимулировать остеопоэз. Лечебная помощь по- неволе сводится к одним лишь симптоматическим мероприятиям. Из них наиболее практически важны влияние на кровотворение с целью противодействия вторичному малокровию, гигиена полости рта во избежение опасной для жизни инфекции и особенно некроза челюсти, а также посильное предотвращение патологических переломов костей. Этим самым может быть улучшено в какой-то мере и общее предсказание.
12. МЕЛОРЕОСТОЗ
Под названием мелореостоз (melorheostosis), а также osteosis eburnisans или osteopathia hyperostotica congenita monomelica описано около 80 случаев врожденного заболевания скелета, выражающегося в совершенно своеобразном одностороннем остеосклерозе костей одной только конечности. Можно не сомневаться в том, что мелореостоз уж совсем не так редко встречается, мы в нашей консультативной практике наблюдали свыше 30 случаев. Вполне естественно, что отдельные новые казуистические наблюдения, не вносящие в науку никаких новых фактов и не возбуждающие новых мыслей и обобщений, остаются неопубликованными.
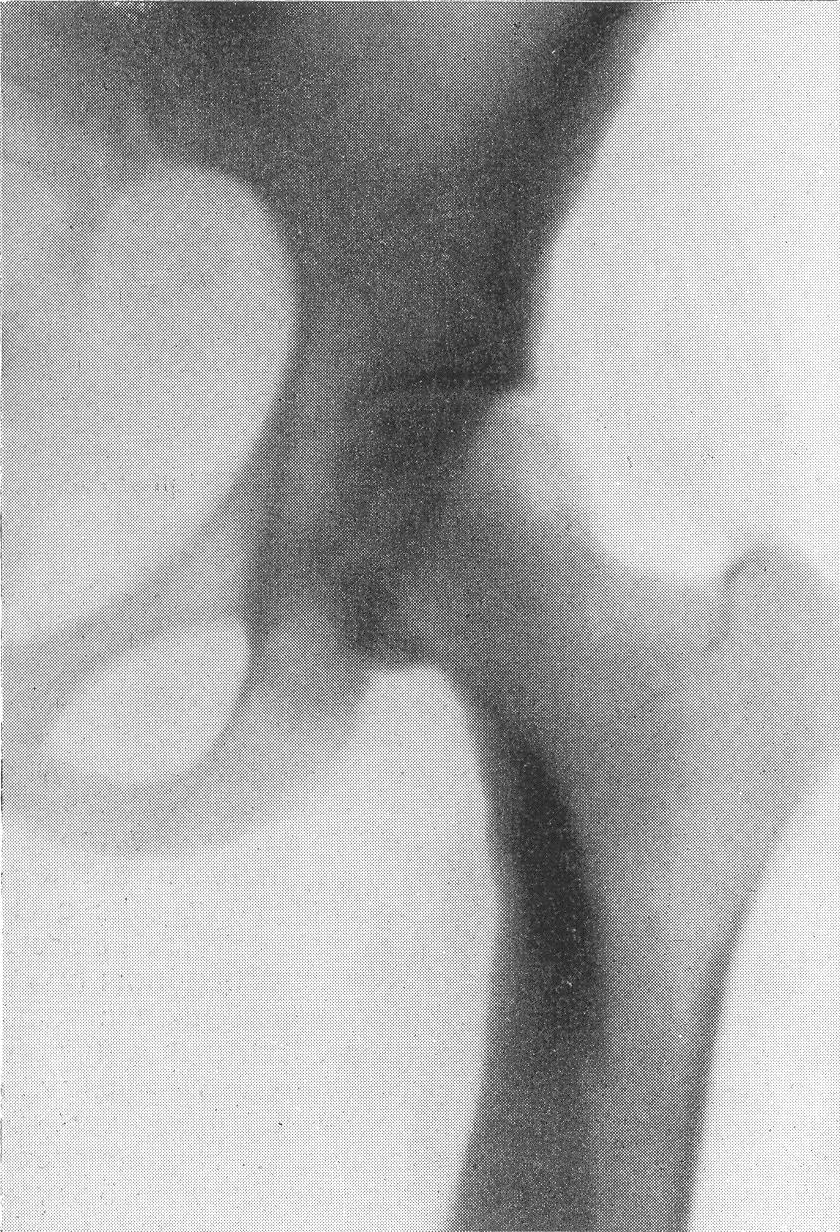{kind=link}
Остеосклероз при этом заболевании захватывает не всю кость по окружности, а простирается слегка волнистой полосой вдоль длинной оси конечности (см. рис. 261, К), переходя через линию суставов на другие кости. Таким образом, может оказаться пораженной целая верхняя или нижняя конечность, или больший или меньший ее периферический участок, например, часть лопатки, полуцилиндр плечевой кости, часть цилиндра лучевой кости и скелет II пальца, вместе с лежащими по этой оси участками запястных костей, или же часть — медиальная (рис. 315) или латеральная — бедренной, большеберцовой кости с продолжением и расширением процесса на предплюсневые и плюсневые кости и соответствующие по длиннику фаланги одного или нескольких, но никогда не всех пальцев (рис. 316). Впервые открывший эту болезнь в 1922 г. совместно с Жоани (Joanny) Лери (Leri) сравнивает остеосклеротические полосы с картиной, напоминающей стекающий со свечи вдоль конечности и застывающий стеарин или воск. Отсюда и название „мелореостоз” — „стекающая вдоль конечности кость” или более точное, но еще более длинное и сложное составное (из пяти корней!) наименование «ризомономелореостоз». Отдельные случаи мелореостоза отличаются друг от друга по степени и форме склеротических участков. Как говорится, случай на случай не приходится, и каждый случай представляет свои неповторяемые иконографические особенности. В более тяжелых случаях поражения нижней конечности захвачена и соответствующая половина таза, включая крестец, при локализации в верхней конечности — часть лопатки, а то и все конечности. Интересно, что из парных костей поражается только одна: это или большеберцовая или малоберцовая кость, или лучевая или локтевая кость (рис. 317), но никогда не обе кости одновременно. В самых редких случаях в какой-то ничтожной степени в процесс может быть вовлечен череп (часть нижней челюсти), а также позвоночник. Поражения ребер мы ни разу не наблюдали. Измененный корковый слой, а также уплотненные эпифизы и мелкие губчатые кости приобретают плотность слоновой кости.
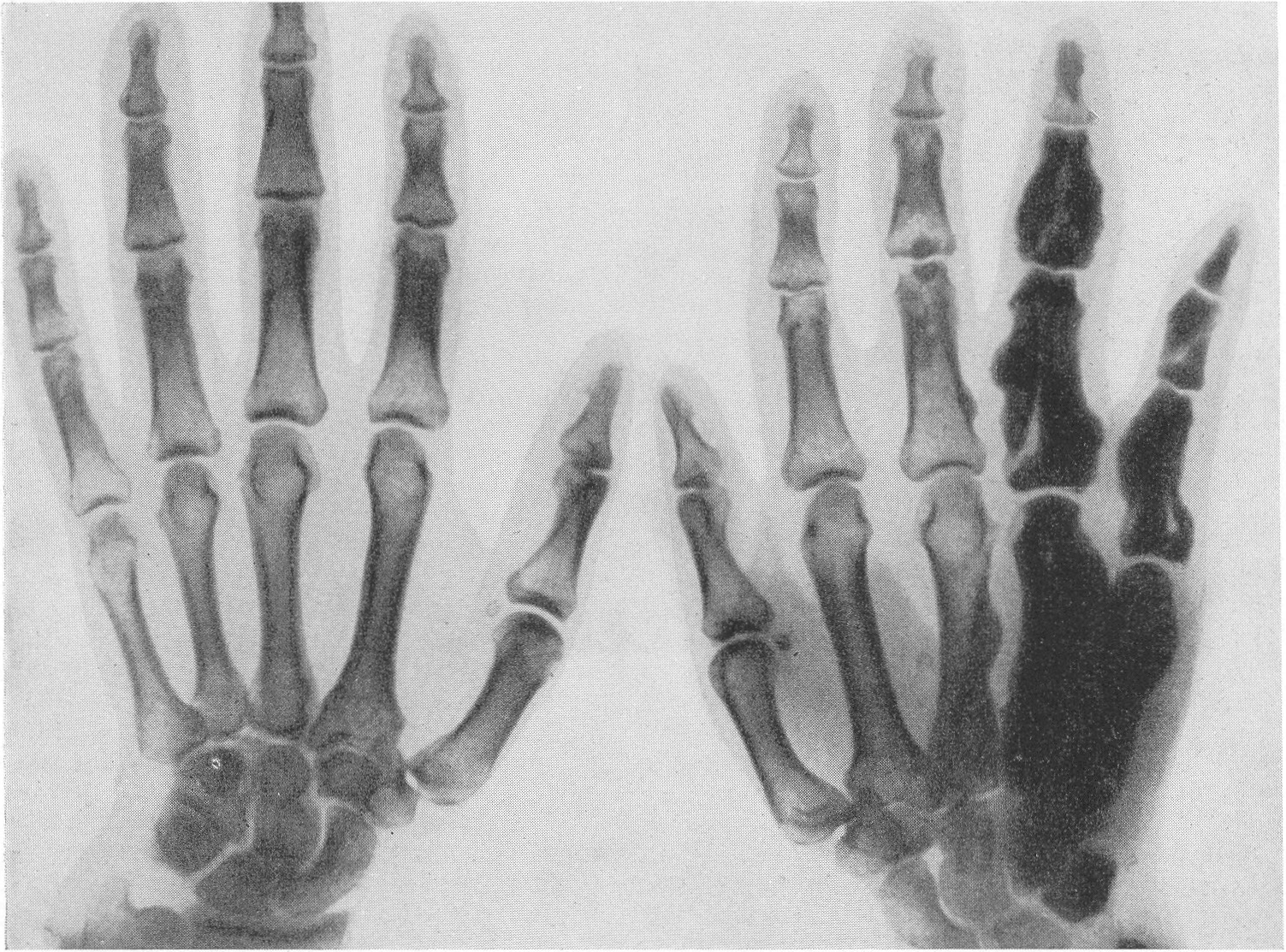{kind=link}
Рис. 316. Мелореостоз Лери у 29-летнего больного. Типичное поражение 21/2 пальцев левой кисти, по ульнарному краю. Девиация IV и особенно V пальца в ульнарную сторону.
Корка утолщается как в сторону костномозгового канала (эндостальная форма), в большей или меньшей степени суживая его, так и наружу (периостальная форма), возвышаясь над нормальным уровнем кости и увеличивая этим ее наружный диаметр. Поверхность „наплывов” слегка волниста, с гребневидными возвышениями и углублениями, но всегда очень резко контурируется. Склеротические полосы и ленты бывают чаще всего сплошными, но нередко они прерывисты, расслоены. Иногда наблюдаются некоторое удлинение костей и их небольшая дугообразная деформация, а подчас и, наоборот, очень незначительное укорочение. Это зависит, надо думать, от участия энхондрального хряща, раздражения или торможения его функции. Костная ткань в соседстве со склеротическими полосами и островами сохраняет нормальный рисунок или часто слегка поротична, так что граница между затемненными склеротическими лентами и костным фоном очень крута. Иногда в мягких частях таза или области плечевого пояса развиваются неправильные шаровидные плотные костные массы, а при прогрессировании болезни подобные островки появляются и в окружности суставов, которые сами по себе особых изменений не представляют. Патологические переломы при мелореостозе не описаны. Мы также по собственному опыту и по литературным данным не знаем озлокачествления при мелореостозе. Течение болезни вообще вполне доброкачественное, и общее предсказание благоприятное. Гистологическое исследование при мелореостозе ничего характерного, кроме склероза пораженных участков, не показывает. Никаких воспалительных изменений в костной ткани не бывает. Уплотненная кость имеет, как и при мраморной болезни, неправильную архитектонику. Надкостница либо вовсе не изменена, либо фиброзно уплотнена. Имеется много незрелого костного вещества, костный мозг атрофичен и также замещен волокнистой соединительной тканью, сосуды находятся в состоянии запустения.
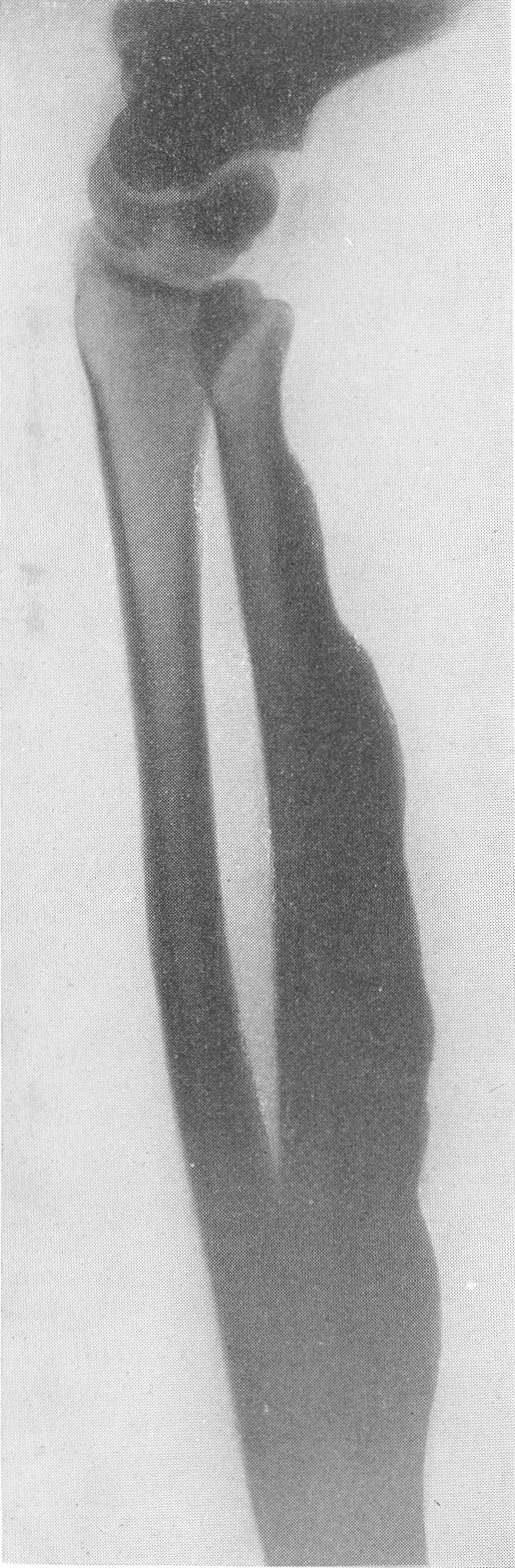{kind=link}
Мелореостоз наблюдается в любом, но чаще всего в детском и юношеском возрасте. В последние годы мы все чаще выявляем эту болезнь и в среднем, и даже в зрелом и старческом возрасте. Клиническая картина по сравнению с рентгенологической относительно очень бедна, но все же рисуется из некоторых симптомов, которые становятся легко понятными в свете рентгенологических данных. В общем чем старше больной мелореостозом, тем больше вступает в свои права клиника, и чем он моложе, тем более над клиникой преобладает рентгенология. Больные чаще всего жалуются на болевые ощущения, характер и интенсивность которых в отдельных случаях колеблются в весьма широких пределах. Боли только очень редко бывают сильными, мучительными. Обычно же это умеренные тупые, глухие боли, усиливающиеся по ночам, подчас зависящие от функциональной активности, даже от времени года. Боли не только связаны по местоположению со склеротическими костными участками, это не только диафизарные боли в больших трубчатых костях, но и боли в области суставов, а также вторичные сосудистые и нервные явления, обусловленные давлением со стороны пышных костных гребней и напластований. Нередко, уже после определения болезни в результате рентгенологического исследования, удается задним числом уточнить, что болевые ощущения так или иначе тревожат пациента годами, пяти- и десятилетиями. Иногда жалобы больных сводятся лишь к чувству усталости в измененной конечности, и ряд наших больных нам указывал без внушения с нашей стороны, что пораженная конечность им кажется более тяжелой, чем противоположная, здоровая. Предъявляются и жалобы на одностороннюю мышечную слабость, и сами мышцы в застарелых случаях оказываются атрофированными и фиброзно уплотненными. Интересно, что в этих же далеко зашедших случаях мелореостоза, соответственно подлежащим костным поражениям, можно отметить также большую или меньшую степень склеродермии. В ряде случаев мелореостоза взрослые больные указывают на тугоподвижность суставов на пораженной стороне. У детей мы этого не видели. С возрастом может развиваться прогрессирующее ограничение подвижности одного или нескольких крупных суставов — все на одной конечности. Объясняются эти мономелические суставные изменения не столько склерозом эпифизарных костных элементов суставов, сколько-все нарастающими отложениями и глыбами гетеротопического костного вещества в мягких тканях в окружности суставов. Это мы наблюдали в области тазобедренного и коленного суставов у больных в уже зрелом возрасте, никогда не у детей. И, наконец, важным внешним клиническим выражением мелореостоза могут служить деформативные явления, из которых раньше всего бросаются в глаза осевые уклонения в сторону (девиации) одного из пальцев кисти. Кровь по своим морфологическим и химическим свойствам при мелореостозе уклонений от нормы не представляет, обмен веществ остается нормальным. При этих условиях нет ничего удивительного в том, что мелореостоз, как правило, длительно протекает скрыто, и точное начало болезни определить обычно невозможно, так как она подкрадывается исподволь. Вполне вероятно, что многие больные и вовсе не распознаются, они привыкают к своим болям и мирятся со своим „хроническим ревматизмом”, не обращаются к врачам, а врачи не обращаются за консультативной помощью к рентгенологии. Точное распознавание мелореостоза возможно исключительно при помощи рентгеновых лучей и на основании рентгенограмм является элементарной задачей, разумеется, при условиях знакомства с причудливой рентгенологической картиной мелореостоза, а также при соблюдении обязательного требования подвергать рентгенологическому исследованию скелет и во всяком случае конечность в целом, а не только ограниченный участок костно-суставной системы, на котором сосредоточено внимание больного и лечащего врача. Эта методическая сторона дела сближает мелореостоз с болезнью Олье, с полиостотической фиброзной остеодисплазией и другими аналогичными мономелическими страданиями. Таким образом, в типичных случаях мелореостоза вряд ли может возникнуть, трудность дифференциально-диагностического порядка, в высшей степени характерная рентгенологическая картина имеет решающее не только устанавливающее, но к дифференциальное диагностическое значение. Ошибки происходят лишь в результате неполноценного клинико-рентгенологического исследования, в недоисследованных случаях. Опыт жизни учит, что по тем или иным клиническим и рентгенологическим соображениям могут потребовать отличительного распознавания гуммозный сифилитический остеопериостит, хронический склерозирующий остеомиелит, травматический периостит, мраморная болезнь, остеопойкилия, деформирующая остеодистрофия Педжета, фиброзная дисплазия костей, склерозирующая остеогенная саркома, неврогенная остеоартропатия, хондроматоз сустава, капсульная остеома и остеосаркома и т. п. Этиология мелореостоза неизвестна. Остается лишь ограничиться общей фразой о том, что это какое-то врожденное эмбриональное метамерное нарушение процесса развития преимущественно костного остова конечности. Это единственная известная нам болезнь, метамерно поражающая кости одной только конечности в виде продольных патологических остеосклеротических полос, к тому же не связанных ни с определенными сосудистыми, ни с нервными источниками. Последнее требует объяснений. Казалось бы проще всего связать удивительную локализацию костных изменений именно и только с иннервационным нарушением. Однако строгий анализ костных поражений верхней, а также нижней конечности не дает определенной связи с тем или иным известным в нормальной анатомии нервным стволом, „наплывы” не соответствуют в отдельных случаях территории разветвлений определенных нервов. И функционально нервы конечности при мелореостозе при исследовании обычными клиническими методами оказываются нормальными. Лечение мелореостоза сводится к симптоматическим мероприятиям.
13. ОСТЕОПОЙКИЛИЯ
Остеопойкилия [от древнегреческого пойкилос, что значит пятнистый, osteopoikilia, или врожденная рассеянная склерозирующая (конденсирующая) остеопатия, osteopathia scleroticans s. condensans disseminata, или врожденная пятнистая множественная остеопатия] впервые описана еще в 1905 г. Штида (A. Stieda), но осталась в литературе как-то незамеченной, пока эту болезнь не „открыли” вновь Альберс-Шенберг, а также Леду-Лебар (Ledoux-Lebard) с сотрудниками. В мировой литературе опубликовано около сотни случаев остеопойкилии, а мы сами ее диагностировали 28 раз. Это врожденное системное заболевание скелета выражается в том, что во всех костях, за исключением, вероятно, ключицы, в губчатом веществе разбросаны более или менее густо сидящие округлые и овальные гомогенные склеротические плотные костные островки величиной от 2 до 8—10 мм (см. рис. 261, Л). Они описаны в любом возрасте, даже у плодов, в первые дни, недели и месяцы жизни, в среднем и пожилом возрасте. Мужской пол поражается немного чаще женского. Наблюдаются как спорадические случаи, так и более редкие семейные, в разных поколениях. Мельник (Melnick) в 1959 г. описывает 17 случаев остеопойкилии, выявленных им рентгенологически у членов одной и той же семьи в 4 поколениях, причем самому молодому было 4 года, а самому старому 74 года. Сильнее всего поражены короткие губчатые кости конечностей (кости запястья и предплюсны), а также эпифизарные концы длинных костей, в меньшей степени усеяны очагами остеосклероза метафизы, а диафизы всегда остаются неизмененными. Обычно поражены головка и шейка бедренной кости, головка плечевой кости, фаланги пальцев рук и ног, пястные и плюсневые кости (рис. 318—320). Исключительно редко склеротические островки располагаются в черепных костях, ребрах. Обычно пощажены и позвонки, только нижние поясничные и крестцовые позвонки могут быть вовлечены вместе с другими костями таза в процесс. Отдельные склеротические пятна подчас удлинены всегда вдоль длинных осей конечности или кости и на полюсах так зазубрены, что органически входят в губчатую структурную сеть. Пораженные участки скелета приобретают при остеопойкилии характерный пятнистый вид; отсюда ведь и название — они кажутся как бы обрызганными кисточкой. В отдельных случаях остеопойкилия может быть мелко- или крупноочаговой, редкой или частой, густой по количеству склеротических островковых элементов. И это не зависит от возраста. Внешняя форма костей всегда остается нормальной. Важно также, что ядра окостенения появляются в нормальные сроки и имеют в процессе их развития нормальную форму и структурный фон. Гистологические исследования Шморля (Schmorl) показали, что островки костного вещества — это не „компактные островки”, т. е. они построены не по типу компактной ткани, а представляют собой сконденсированную, густотрабекулярную сеть губчатого вещества, без каких-либо других микроскопических особенностей. На периферии кости островки сливаются с компактным корковым веществом. Эволюция островков и их судьба в зависимости от возраста и других факторов недостаточно убедительно прослежена. Имеются указания на возможность их уменьшения и даже исчезновения. Структурные изменения скелета при остеопойкилии не сопровождаются особыми объективными или субъективными клиническими проявлениями. Только в последние годы, с накоплением коллективного опыта, удалось выяснить факт серьезного принципиального значения, что в некоторых случаях, преимущественно у взрослых, остеопойкилия сочетается со своеобразным, до известной степени аналогичным кожным заболеванием, а именно рассеянным чечевицеподобным кожным фиброзом (dermatofibrosis lenticularis disseminata). Очевидно, что в основе изменений в обеих системах — костной и кожной — лежит какое-то иннервационное, нервнотрофическое нарушение. Поскольку остеопойкилия не связана с определенными внешними клиническими проявлениями, ясно, что это заболевание распознается только при рентгенологическом исследовании, произведенном по какому-либо другому поводу, как говорится, случайно.
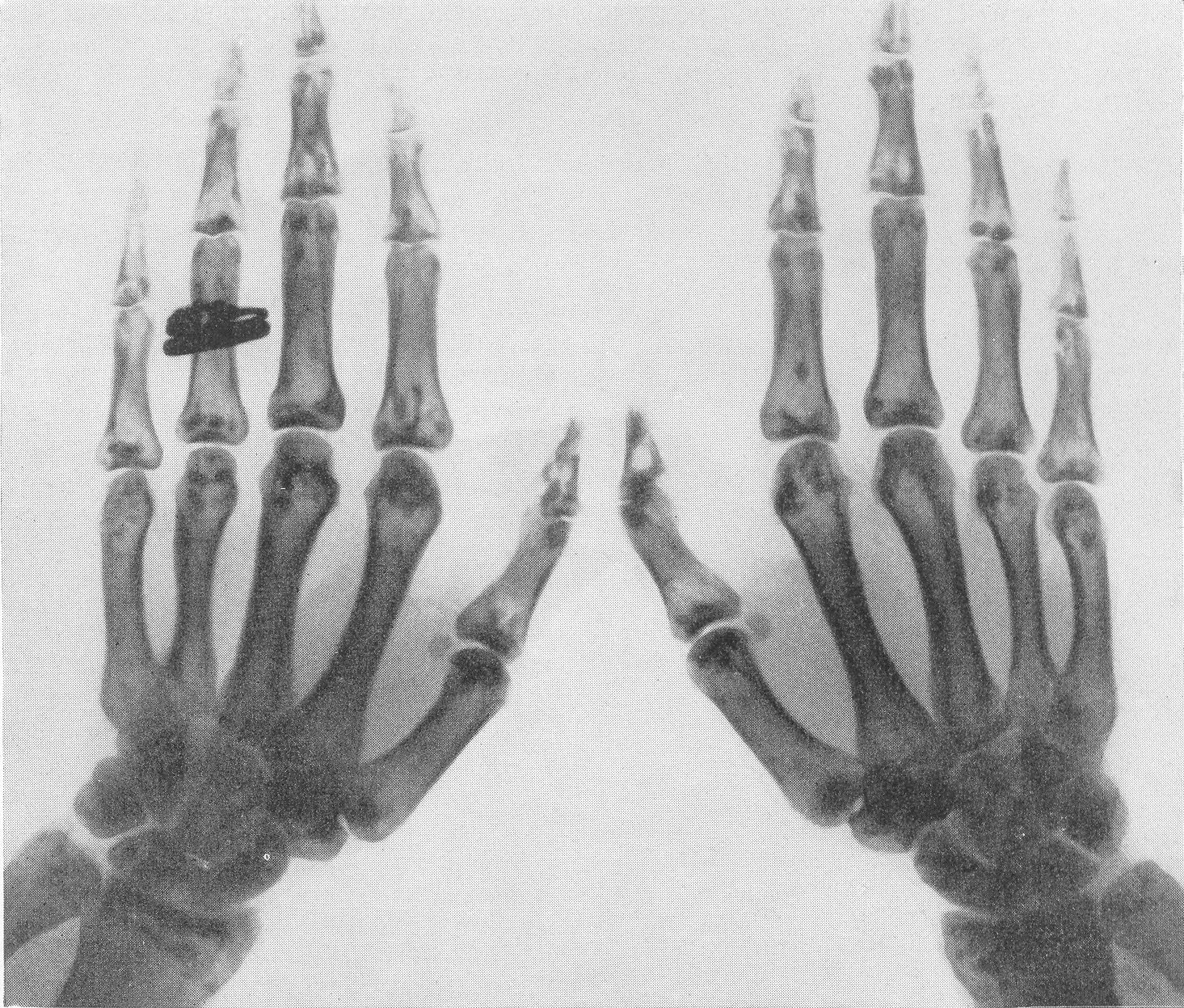{kind=link}
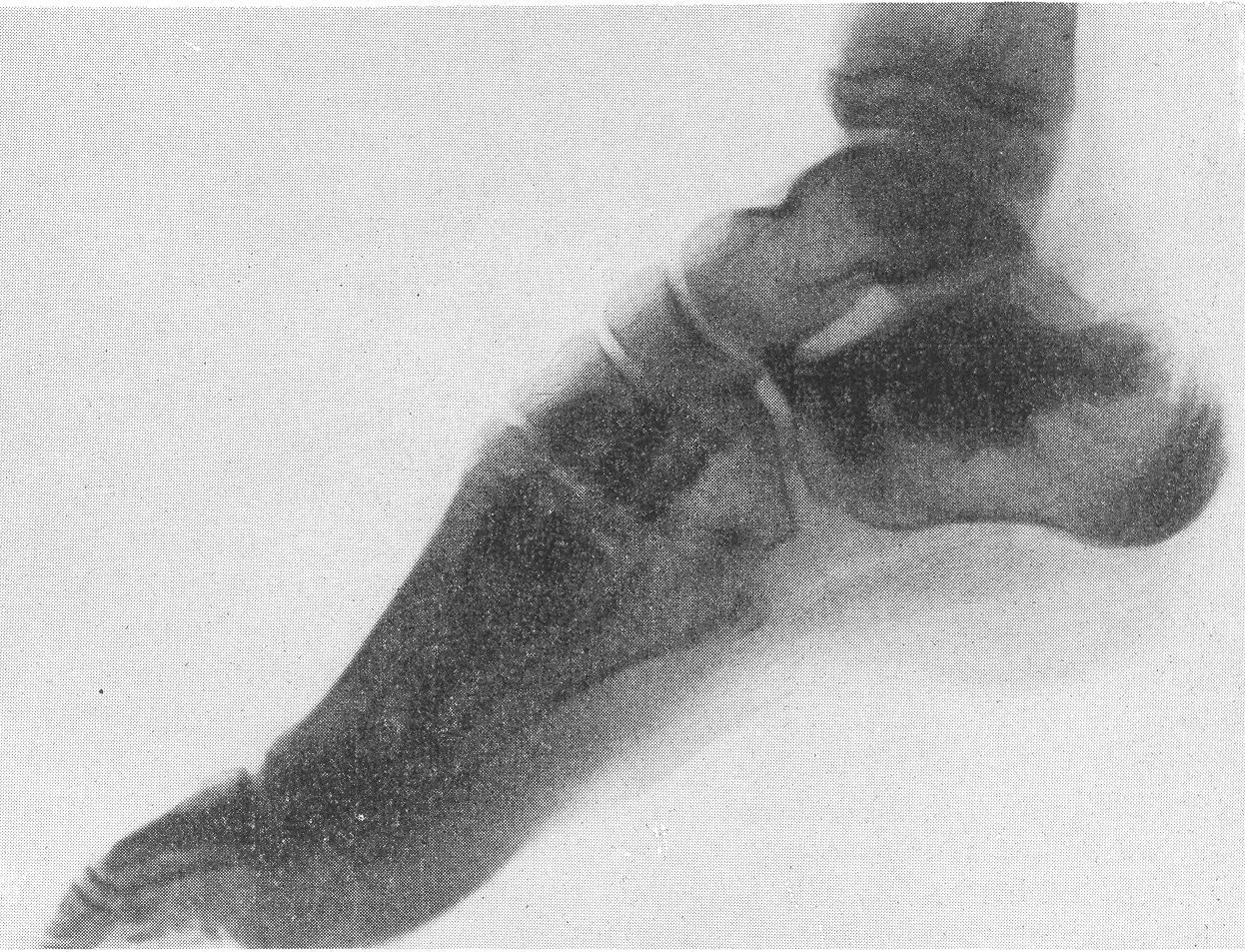{kind=link}
В высшей степени типичная рентгенологическая картина остеопойкилии делает дифференциальную диагностику излишней. Единственная болезнь, с которой еще возможно смешение остеопойкилии, это ниже описываемая врожденная эпифизарная точечная костная дисплазия. Ворхув (Voorhoeve) опубликовал несколько случаев врожденной структурной аномалии скелета, объединяющей некоторые черты и мраморной болезни, и мелореостоза, и остеопойкилии. Этот автор наблюдал на рентгенограммах метафизов длинных трубчатых костей и также в других отделах скелета множественные продольные параллельные друг другу или лучисто расходящиеся склеротические полосы. Кроме того, в пределах губчатой сети располагались отдельные изолированные островки остеосклероза. Метафизы были слегка увеличены в размерах, и на их поверхности находились отдельные возвышения и шероховатости.
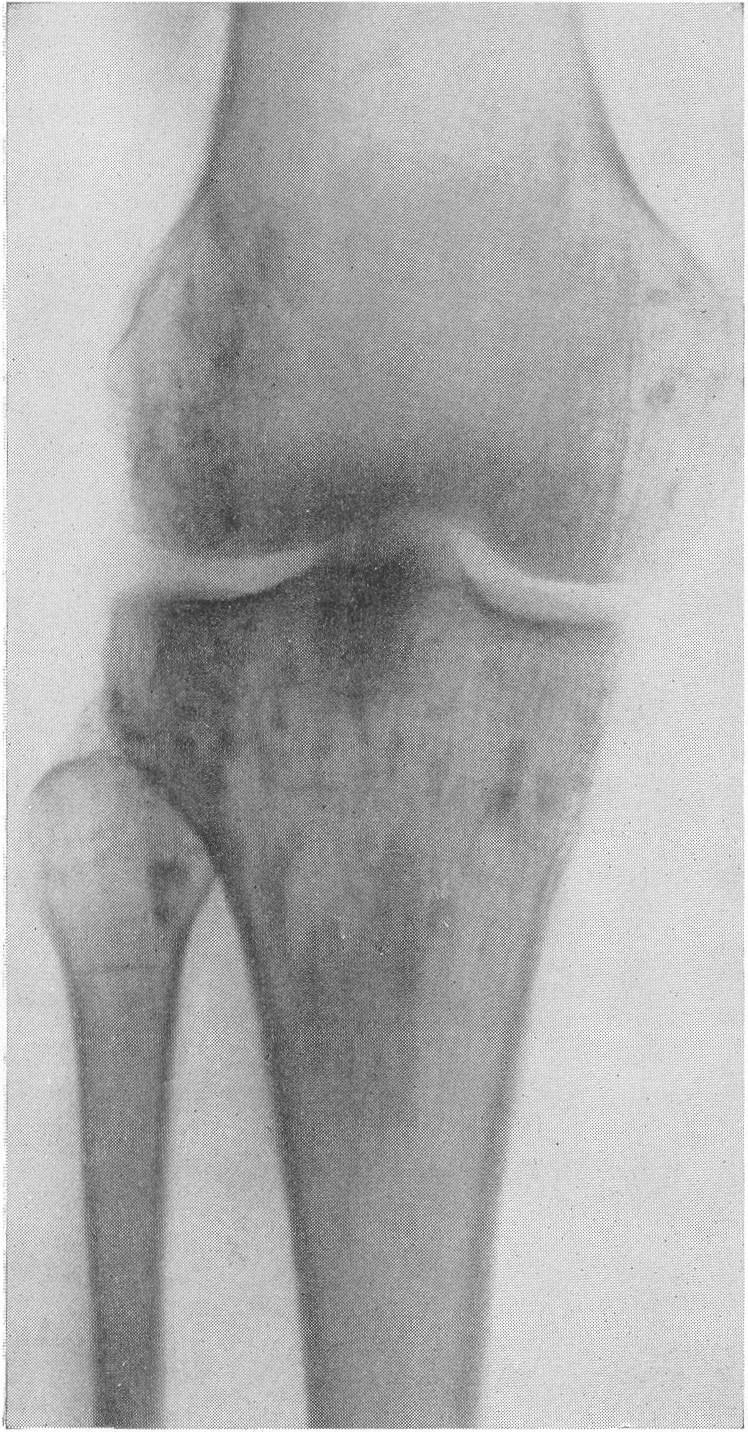{kind=link}
14. АРАХНОДАКТИЛИЯ
Арахнодактилия описана в литературе впервые в 1896 г. известным французским педиатром Марфаном (Marfan) под названием долихостеномелия и до сих пор еще сплошь и рядом обозначается как болезнь Марфана. Настоящее же общепринятое наименование „арахнодактилия” предложил в 1902 г. Ашар (Achard). Это врожденное системное заболевание относится к числу сравнительно редких. Как и другие представители этой группы болезней, арахнодактилия встречается то неожиданно спорадически, то в качестве семейного заболевания, которому подвержено несколько членов одной и той же семьи из одного или разных поколений. Преимущественного поражения мужского или женского пола и здесь никто не отметил. Арахнодактилия выражается в непомерном удлинении всех трубчатых костей, в особенности же наиболее периферически расположенных — фаланг рук и ног, пястных и плюсневых костей, но также и в изменениях черепа, скелета туловища. Ручки и ножки становятся поразительно длинными и узкими, вытянутыми вдоль длинных осей, и пальчики согнуты в тонких суставчиках. Это действительно порождает подобие с члениками паука, а отсюда и удачное, пусть и чисто внешнеописательное, название болезни — арахнодактилия (от древнегреческого арахнос — паук). Кроме изменений опорно-двигательного аппарата имеются еще обязательно очень характерные глубокие поражения других систем, в первую очередь изменения глаз и сердечно-сосудистые, что опять-таки говорит о том, что все врожденные системные так называемые костные болезни никоим образом не являются одним только костным патологическим процессом, а лишь костным или преимущественно костным выражением общего заболевания человеческого организма. Патологический процесс при арахнодактшши начинается, несомненно, в ранней стадии внутриутробного развития и, стало быть, всегда уже выражен у новорожденного ребенка и в первый период жизни. Но совсем нередко болезнь проявляется и усиливается лишь при дальнейшем росте и развитии ребенка. Арахнодактилия — очень тяжелая болезнь с весьма неблагоприятным предсказанием, т. е. с очень высокой общей летальностью, и большинство детей гибнет в первые же годы жизни от любой привходящей, для них всегда опасной болезни. Лишь очень редко больные арахнодактилией остаются в живых до старшего или юношеского возраста, а тем более до зрелости и даже старости, и то это относится лишь к относительно весьма редким ее доброкачественным формам. К обязательным симптомам арахнодактилии следует причислить прежде всего резко выраженную общую мышечную атрофию с атонией всей скелетной мускулатуры, типа врожденной амиотонии [myatonia congenita Оппенгейма (Oppenheim)]. Мы никогда ни при одном другом заболевании не видели ничего, отдаленно приближающегося к состоянию мышц у больных арахнодактилией. Мышцы конечностей на ощупь, можно сказать, просто как бы отсутствуют, они вообще не выделяются из-под кожи, они имеют в тяжелых случаях этого заболевания консистенцию буквально слизи. Это впечатление усугубляется еще тем обстоятельством, что кожа крайне истончена, а жировой клетчатки у страдающих арахнодактилией характерным образом вообще не бывает, она не развивается. Между суставами, например локтевыми, коленными, но уж во всяком случае между пальчиками мы нередко находим тонкие полупрозрачные кожные перепонки, наподобие плавательных мембран у водоплавающих птиц. Кроме недоразвития мускулатуры и подкожного жира имеется еще крайняя степень дряблости связочного аппарата, так что отдельные длинные тонкие членики конечностей чрезмерно пассивно подвижны. Самостоятельные движения детей, страдающих арахнодактилией в тяжелой форме, почти вовсе не бывают или они сведены к минимуму движений некоторых групп мышц. Это крайне ограниченные по объему, замедленные, какие-то неживые, можно сказать, нечеловеческие повороты головы из стороны в сторону. Поэтому эти больные несчастные дети лежат пластом, прикованы к кровати и целиком зависят от обслуживающего персонала. Из-за недоразвития костей, мышц, мезенхимальных образований и жира общий вес тела больных арахнодактилией очень мал. Поразительна общая слабость, и даже в самых благоприятно протекающих случаях у выживающих наступает утомляемость даже после незначительных физических усилий. Таким образом, налицо всегда обязательная и резко выраженная недостаточность статических и механических функций опорно-двигательного аппарата в целом. При осмотре больного арахнодактилией производит неизгладимое впечатление еще внешний облик личика ребенка. Голова большая, череп увеличен и вытянут в длину, имеется долихоцефалия. Постоянным признаком, вероятно, обусловленным неподвижным лежанием на спинке, является уплощение затылочной области, а также выступление вперед лобных бугров, лоб всегда высокий, крутой. Корень носа широк и погружен. Авторы указывают на изменения челюстей: то это выпячивание нижней челюсти вперед, прогнатия, то это, наоборот, срезанный подбородочек, что придает личику что-то напоминающее птицу. Вообще выражение лица у детей с арахнодактилией не детское, а старческое, скованное, грустное, меланхолическое. Очень важны для общей клинической характеристики арахнодактилии постоянно выраженные многочисленные и разнообразные неправильности развития глаз. Глаза широко расставлены (гипертелоризм) и лежат глубоко под нависшими над ними выдающимися надбровными дугами. В 30—50% всех случаев арахнодактилии имеется двусторонний симметричный подвывих или вывих хрусталика, смещенного именно кверху. Передняя камера глаза плоска, имеется близорукость. Сетчатка пигментирована. Зрачки узки и при движениях глаз появляется мелкое дрожание радужек (иридодонез). Вывихнутые хрусталики часто мутнеют, что еще в большей степени ухудшает зрение, и обычно с общей неподвижностью тщедушного тельца гармонирует неподвижный невидящий взгляд больного. Еще чаще, чем поражения глаз, у арахнодактиликов наблюдается неправильное развитие ушных раковин. Они очень велики, не содержат хрящей, а потому мягки, дряблы, лапчаты, легко подвертываются под головку или щечку при боковых движениях. Волосики на голове скудны, сухи и жестки. Часто, почти в половине всех случаев, определяется и та или иная неправильность, развития сердца, главным образом мышцы его, но нередко и клапанов, перегородки между камерами, отхождения крупных сосудов, т. е. врожденный порок сердца. В последние годы хорошо рентгенологически изучены [особенно при помощи контрастных методов исследования Стайнбергом (Steinberg)] весьма характерные для арахнодактилии аневризматические расширения аортальных бульбарных синусов, расположенных непосредственно над аортальным артериальным кольцом, подчас огромных уродливых размеров.
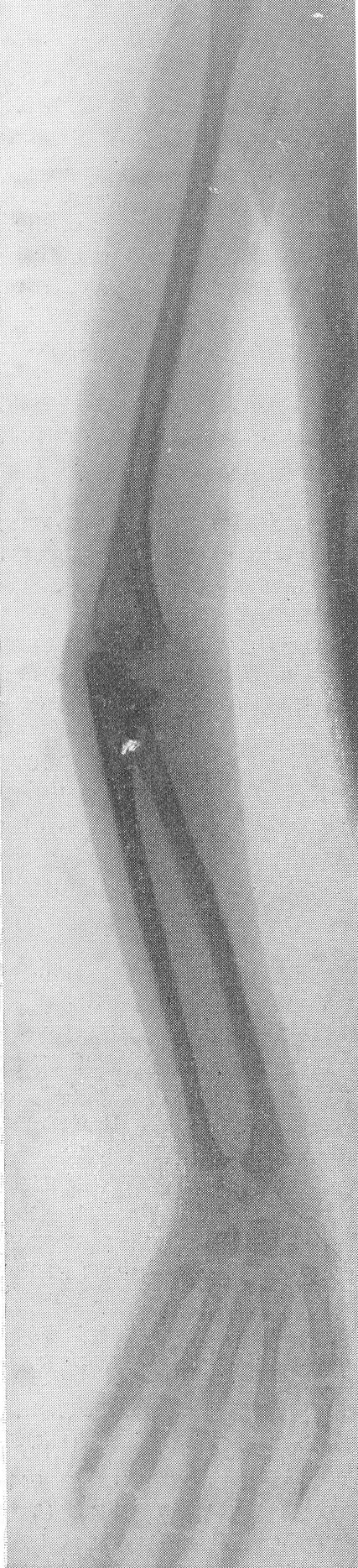{kind=link}
Рис. 321. Арахнодактилия. Рентгенограмма верхней конечности у 4-летней девочки с резко выраженной клинико-рентгенологической картиной.
Центральная нервная система всегда при арахнодактилии изменена, имеется резкая общая отсталость умственного развития. Явных эндокринных нарушений не бывает. В частности, много внимания уделено исследователями в литературе, да и нами в наших собственных наблюдениях, гипофизарной системе. Но убедительных данных в пользу поражения гипофиза, в особенности его передней доли, мы не получили. И в многочисленных случаях вскрытий, опубликованных в литературе, передняя доля гипофиза, как и все исследованные звенья эндокринной цепи, неизменно оказывались без четких патологических изменений. Определенных биохимических сдвигов крови при арахнодактилии никто еще не нашел. В частности, изменений кальция и фосфора сыворотки крови, несмотря на всеобщее поражение костной системы, мы ни разу не обнаружили. Этиология арахнодактилии неизвестна. Поэтому мы ограничиваемся лишь общим неконкретным замечанием о том, что арахнодактилия — это какой-то врожденный глубокий порок развития по преимуществу мезодермальной системы (глаза-то ведь эктодермального происхождения!), какая-то глубинная врожденная мезенхимальная дистрофия. В последние годы вырабатывается, по-видимому, более точное представление о том, что в основе болезни лежит генерализованное поражение эластической арматуры во всем организме, специфическое недоразвитие эластической ткани, где бы она ни располагалась, своеобразный врожденный системный эластоз. Этот порок вызывается пока еще неизвестной нам причиной уже в очень ранней стадии эмбрионального развития. Распознавание арахнодактилии при условии достаточного знакомства с этой болезненной нозологической формой не представляет никаких трудностей. Рентгенологическое исследование вносит дополнительно к клиническим данным много ценных подробностей, обогащающих наши знания об этой болезни. Ведущим рентгенологическим признаком арахнодактилии является диспропорция в форме каждой исследованной трубчатой кости, как большой, так и малой (см. рис. 261, Е). До крайности, каррикатурно, как в кривом (цилиндрическом) зеркале, преувеличены длинные размеры и преуменьшены поперечники костей (рис. 321). Арахнодактилия — это диаметральная противоположность хондродистрофии. Кости грацильны, удлинены и истончены, и как уже сказано, это особенно касается фаланг, пястных и плюсневых костей. Большинство авторов при этом указывает на не только относительное, но и абсолютное удлинение конечностей, т. е. истинное увеличение роста тела в длину, которое усиливается с течением времени у тех больных, которые выживают. В школьном возрасте, например, рост больных опережает таковой их ровесников, служащих для контроля, на ряд лет. Интересно, что при крайнем удлинении и истончении костей они не искривляются. Удивительно также, что арахнодактилии совершенно не свойственны патологические переломы, и мы их можем видеть только по вине неосторожного медицинского персонала. Общий структурный рисунок костей остается довольно правильным. Имеется лишь системная атрофия костей с остеопорозом, рарефикация всего костного вещества, чему, очевидно, в немалой степени способствует общая обездвиженность больных. Корковый слой тонок, а в губчатом веществе трабекулярная сеть редка, трабекул мало, а отдельные трабекулы истончены. Это полностью подтверждается многочисленными гистологическими исследованиями. Мышечные бугры и возвышения, как и эпифизарные концы костей, повсеместно выражены слабо. Наблюдаются псевдоэпифизы. Темпы окостенения могут быть в единичных случаях извращены, обычно ускорены. В клинико-рентгенологической картине скелета при арахнодактилии большую роль для формирования общего вида больного имеют еще многочисленные, по существу вторичные деформации. Сюда относятся всегда резко выраженные плоские стопы, нередко конские стопы и тому подобные деформативные процессы, genua valga и recurvata, coxa valga. Слабость мягких тканей суставов, даже без увеличения мышечной тяги, влечет за собой разболтанность больших и малых суставов, подвывихи и резкие вывихи с порочными положениями конечностей. Так, например, мы наблюдали вывихи в суставах больших пальцев стоп, в плечевых и в тазобедренных суставах. В одном нашем случае был асимметричный односторонний вывих бедра, в результате которого бедро легло рядом с краем туловища и колено очутилось в подмышечной впадине! Подчас вызывает удивление крайне подвижная и смещенная далеко в сторону надколенная чашечка. Грудная клетка обычно резко деформирована, она чаще всего приобретает форму так называемого паралитического или воронкообразного thorax с вдавленной наподобие корыта грудиной. Мы видели однажды весьма странную картину резко сплющенной спереди назад и раздавшейся в стороны, на редкость плоской и широкой, как бы пропущенной через ролики вальца, грудной клетки. Деформация может еще больше осложниться, если присоединяются те или иные искривления позвоночника — чаще всего сколиозы или кифосколиозы, причем отдельные позвонки никаких специальных изменений не представляют. Обезображивается, естественно, и таз, также сплющенный спереди назад. Арахнодактилию практически ни с какой другой болезнью смешать нельзя, и дифференциальной диагностики ее и не существует, так как в выраженных случаях все клинические и рентгенологические проявления этой болезни сами по себе слишком показательны. Ошибки могут возникнуть только в случае слишком необоснованного расширительного толкования понятия арахнодактилии, т. е. слишком вольной диагностики арахнодактилии у истощенных, крайне худых хронических больных высокого роста. Спор решает наличие, помимо костных изменений, перечисленных выше, характерных симптомов вовлечения в общий процесс других систем и органов в их оригинальном и неповторяемом сочетании. Вряд ли также возникнет в практической жизни надобность в проведении отличительного распознавания между арахнодактилией и гигантизмом любого другого происхождения, например гипофизарного, скажем, при акромегалии, так как, кроме общего признака удлинения конечностей, здесь больше ничего сходного нет. И по отношению к арахнодактилии приходится сделать вынужденное признание, что лечить эту болезнь мы в настоящее время в корне не умеем.
15. СИНДРОМ ЭЛЕРСА—ДАНЛО
Синдром Элерса—Данло (Ehlers, Danlos) — это редкая, этиологически неясная, но весьма определенная и клинически характерная врожденная семейная болезнь, известная с начала нашего столетия главным образом в дерматологии. Ее выражением является следующая пентада: гиперэластоз кожи — чрезвычайно повышенная эластичность кожи (cutis hyperelastica), большая ранимость кожи и сосудов, чрезмерная подвижность суставов, образование особых припухлостей над костными возвышениями и, наконец, множество узлов („сферул”) в коже. Мягкая бархатистая кожа рыхло связана с подкожными слоями, крайне растяжима, как у щенят, — взятая пальцами врача в складку, она может быть приподнята чуть ли не на 10 см, но, будучи отпущенной, она сразу же сглаживается. Малейшее травматическое повреждение кожи создает обширную зияющую рану, которая излечивается при помощи грубого линейного рубца. Суставы настолько подвижны, что кончики пальцев могут быть сближены к тылу кисти. Нередко бывают еще другие, но не столь обязательные аномалии. Рентгенологически сами кости и суставы обычно все же существенных уклонений от нормы не представляют. Наблюдаются костные сращения проксимальных концов луча и локтя, радиоульнарный синостоз, косолапость, замедление процессов энхондрального окостенения, неправильности развития черепа, аномалии зубов. Но главный рентгенологический интерес вызывается при синдроме Элерса—Данло тем обстоятельством, что множественные узлы в коже, обусловленные кровоизлияниями из-за ломкости сосудов и некроза жировых комков, обызвествляются в виде кольцевых или однородных, но не слоистых теней на конечностях и очень напоминают ангиолиты. Дифференциальной рентгенодиагностике помогает анализ числа, расположения и структуры обызвествленных шариков: густое поверхностное внутрикожное их местоположение при синдроме Элерса—Данло и более глубокое, соответствующее определенной сосудистой территории при ангиолитах, дающих слоистые разнообразные по величине затемнения при гемангиомах и ангиоматозе, при флебитах и флебэктазиях. Совершенно различна клиническая картина при обоих заболеваниях.
16. ВРОЖДЕННЫ ЧЕРЕПНО-ЛИЦЕВОЙ ДИЗОСТОЗ
Врожденный черепно-лицевой дизостоз (dysostosis craniofacialis congenita), известный издревле, впервые научно описан в 1912 г. Крузоном (Crouzon) и в литературе многими и обозначается как болезнь Крузона. Это одно из редких врожденных системных заболеваний с невыясненной этиологией, отличающееся весьма постоянными и крайне яркими внешними проявлениями. Фактор семейный при болезни Крузона выражен в сильной степени, это обычно целые поколения одинаково обезображенных болезнью людей, в остальном ничем не отличающихся от вполне нормальных. Однако известен также вариант несемейный и ненаследственный, т. е. спорадический. Черепно-лицевой дизостоз определяется тремя группами основных признаков, а именно характерными изменениями мозгового черепа, лицевого черепа и глаз. Все это в буквальном смысле этих слов внешне видимые уклонения от нормы, и так как болезнь придает всем своим жертвам чрезвычайно типичный внешний облик и все страдающие болезнью Крузона удивительно друг на друга похожи, диагноз заболевания можно уверенно ставить при первом взгляде на больного. В чем заключается это порожденное болезнью единообразие людей, страдающих черепно-лицевым дизостозом? Укажем здесь на клиническую и рентгенологическую семиотическую картину, не разделяя внешнюю и скиалогическую симптоматологию. Мозговой череп имеет почти правильную шаровидную форму или скорее всего оксицефаличен. Его размеры немного уменьшены. Налицо краниостеноз, все швы облитерированы, зарощены. Кости свода черепа резко истончены. Повсеместно выделяются резко углубленные пальцевые вдавления. В области свода черепа у большого родничка имеется некоторое легкое симметричное справа и слева от срединной линии выпячивание костных покровов. Основание черепа при этой разновидности краниостеноза укорочено и углублено. Турецкое седло в связи с этим располагается почти вертикально. Глазницы значительно уплощены. Поэтому глазные яблоки сильно выжаты вперед, имеется резкое пучеглазие. Этот экзофтальм типичен и своеобразен: глаза выпячены не только вперед, но и в стороны, они расходятся. Нередко бывает атрофия зрительных нервов и стало быть понижение зрения. Верхняя челюсть и носовые кости недоразвиты, атрофичны. Нижняя же челюсть значительно выступает вперед (прогнатия). Из-за этого маленький нос сидит глубоко и получает ни с чем не смешиваемую крючковатую форму — пресловутый для черепно-лицевого дизостоза „клюв попугая”. Смешивать этот семейный краниофациальный дизостоз можно разве только с другими дизостозами, например с ниже описываемым черепно-ключичным, да и то скорее по названию, чем по существу. При отличительном распознавании следует также учесть, что в отдельных случаях заболевания и в особенности в отдельных пораженных семьях могут наблюдаться стойкие сочетания ряда врожденных неправильностей, несколько выходящие из указанных выше типичных рамок. Такова, например, комбинация признаков черепно-лицевого дизостоза с более резко выраженной окси- или акроцефалией, аномалиями зубов, умственной отсталостью и сращениями пальцев на руках и ногах, которая в литературе называется акроцефалосиндактилией, или болезнью Апера (Apert). Это только разновидность типичного черепно-лицевого дизостоза.
17. ВРОЖДЕННЫЙ ЧЕРЕПНО-КЛЮЧИЧНЫЙ ДИЗОСТОЗ
Врожденный черепно-ключичный дизостоз (dysostosis cleidocranialis congenita) также известен давно. Его клиническую картину впервые описалв 1883 г. Барлоу (Barlow). Но особенно подробно эта болезнь изучена Пьером Мари и Полем Сентоном (Pierre Marie et Paul Sainton) в 1897 г. Этот дизостоз встречается значительно чаще дизостоза черепно-лицевого, и в мировой литературе описано не меньше 400 случаев этого заболевания. Оно также имеет ярко выраженный семейный характер. По вопросу об этиологии и патогенезе можно не повторяться — они остаются неразгаданными. При врожденном черепно-ключичном дизостозе костная патология гораздо более богата, нежели при черепно-лицевом его собрате, она распространяется на более обширные отделы скелета, гораздо более многообразна, т. е. может быть вовлечен в процесс в большей или меньшей степени почти весь скелет. Но наиболее значительными и показательными являются уклонения от нормы со стороны черепа и ключицы, что и выражается в общепринятом названии.
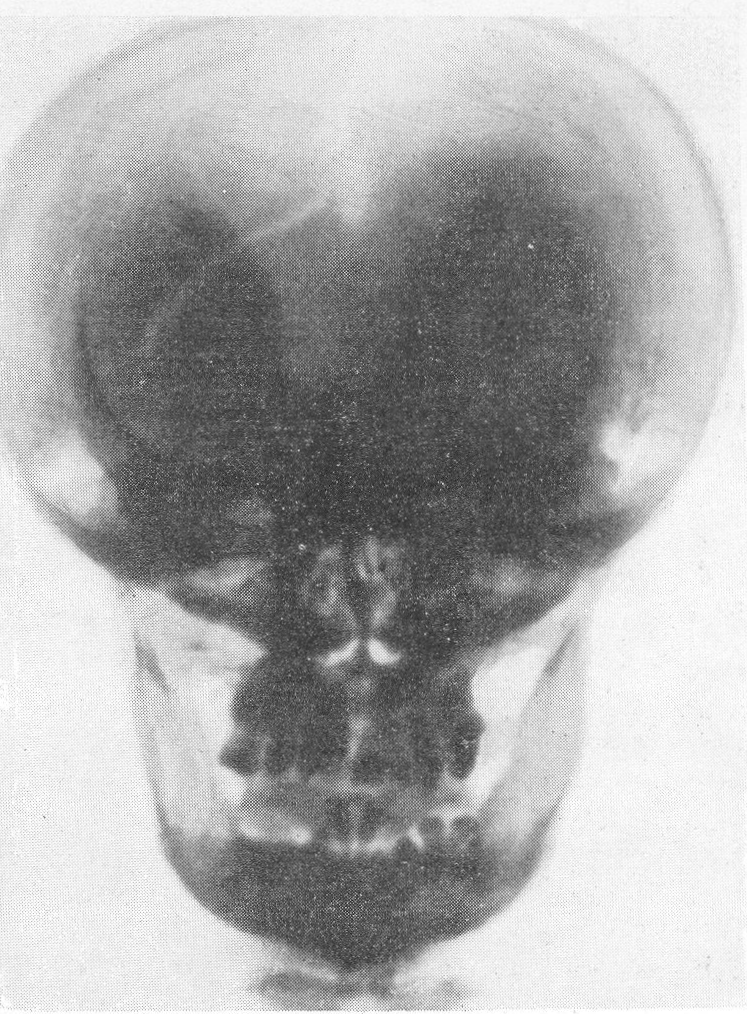{kind=link}
Наиболее характерным клиническим проявлением черепно-ключичного дизостоза служат специфические изменения ключиц — их врожденные изъяны. Больной в состоянии сблизить по средней линии кпереди от туловища оба плеча таким образом, что они полностью соприкасаются друг с другом. Кроме того, из-за изменений черепа, а именно увеличения мозговой части и уменьшения лицевой, больной принимает также своеобразный внешний облик, имеется нарушение пропорции между большим и широким лбом и маленьким лицом. Из важных рентгенологических деталей укажем прежде всего на то, что мозговой череп заметно увеличен именно в поперечнике, а в переднезаднем диаметре он уменьшен, имеется брахицефалия (рис. 322). Выпуклые части свода черепа несоразмерно выдаются и возвышаются. Наблюдаются изъяны свода черепа на местах перекреста швов — своеобразные фонтанеллы, и большой родничок может оставаться открытым в течение всей жизни. Обычно развиваются и многочисленные отдельные добавочные костные включения среди швов — вормиевы косточки. Глазницы же при черепно-ключичном дизостозе в противовес дизостозу черепно-лицевому не представляют уклонений от нормы, а потому и глаза обычны. Основание черепа остается нормальным или же в какой-то небольшой мере недоразвито. Зато и при этом дизостозе кости лицевого черепа, особенно верхние челюсти с их придаточными полостями, недоразвиты, малы. Твердое небо укорочено. А так как нижняя челюсть при этом сохраняет нормальные размеры, то и здесь нередко развивается прогнатия. Недоразвитие лицевого черепа может быть иногда асимметричным. Характерным для черепно-ключичного дизостоза следует считать запоздалое и несовершенное развитие постоянных зубов, и в челюстях надолго, например до 25—30-летнего возраста, задерживаются молочные зубы. Важно подчеркнуть, что больные этим дизостозом и многообразной костной патологией отличаются общим хорошим состоянием здоровья и нередко целыми семьями работают цирковыми артистами. Они попадают к нам и вообще становятся объектом медицинского интереса сплошь и рядом из-за многих разнообразных жалоб именно на свои зубы. Об этом должны знать врачи-стоматологи, чтобы из-за частного не упустить общее и, пожалуй, главное. Решающим и ведущим рентгенологическим симптомом клейдокраниального дизостоза служат дефекты ключиц (рис. 323). Полное отсутствие всей ключицы — это редкость. Обычно отсутствует наружный (акромиальный) конец ключицы, в то время как внутренний (стернальный) конец налицо. Но иногда ключица состоит и из двух фрагментов. Латеральный конец более длинного грудинного отрезка ключицы может быть закруглен, он обычно из-за мышечной тяги приподнят кверху и кпереди. Акромиальный же конец также закруглен, гладок и направлен книзу и кзади. В редких случаях отсутствует как раз стернальный конец. Длина дефекта различна. Костные концы соединены фиброзным тяжем. Бывают стало быть самые различные варианты частичной агенезии или гипоплазии ключиц. Неправильность развития ключицы может повлечь за собой сдавление плечевого нервного сплетения, а также общую мышечную слабость соответственно верхним конечностям.
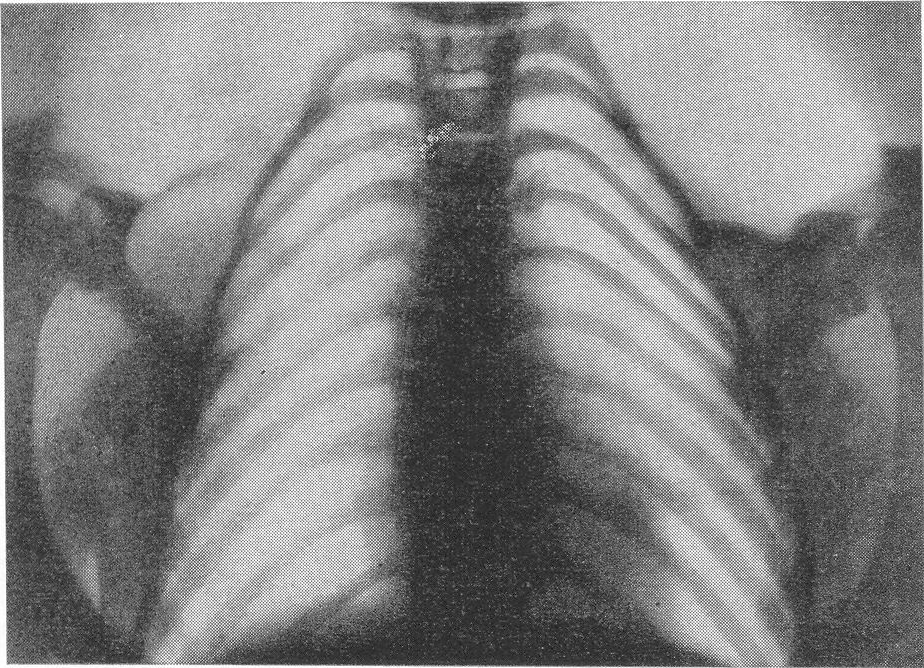{kind=link}
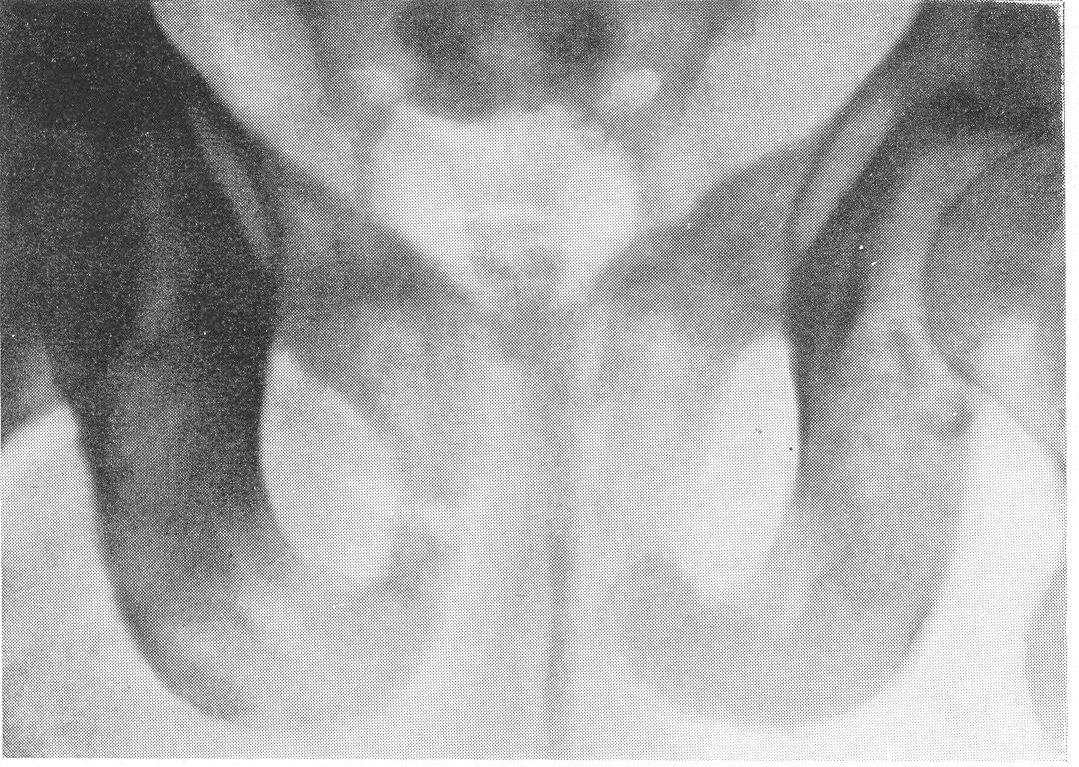{kind=link}
Рис. 324. Врожденный черепно-ключичный дизостоз у той же больной. Агенезия симфиза лонных костей из-за недоразвития последних.
При резко выраженных явлениях сдавления нервных стволов костными или плотными фиброзными элементами вполне целесообразно хирургическое вмешательство. Наконец, выявляются рентгенологически и другие разнообразные неправильности развития по преимуществу не только костей соединительнотканного, но и хрящевого происхождения, говорящие об общей задержке и качественных изменениях процессов окостенения. Сюда относятся в первую очередь поражения шейки бедра со вторичной варусной деформацией, уменьшение размеров лопатки, лонных костей и других элементов таза, в частности крестцовой кости, с неслиянием соответствующих отделов, например агенезией симфиза (рис. 324) и т. д. В некоторых случаях отмечены изменения фаланг — укорочения дистальных фаланг, особенно первых пальцев рук и ног с отсутствием ногтевого бугорка, а также непропорционально длинные II пястные и плюсневые кости; кости голени и предплечья всегда нормальны. Иногда не срастаются дужки позвонков, в то время как тела их патологических сдвигов не обнаруживают, а только несколько усиливаются нормальные кривизны позвоночного столба. При тщательном клиническом и общем рентгенологическом исследовании всего скелета, а не только местных костных неправильностей, которые по той или иной причине обращают на себя внимание самого больного и лечащего врача, всегда при черепноключичном дизостозе можно и должно прийти к правильному устанавливающему и дифференциально-диагностическому заключению.
18. ВРОЖДЕННЫЙ ЧЕЛЮСТНО-ЛИЦЕВОЙ ДИЗОСТОЗ
Врожденный челюстно-лицевой дизостоз (dysostosis mandibulo-facialis congenita) впервые описан в 1889 г. английским офтальмологом Берри (Berry) и еще более обстоятельно в 1944 г. швейцарским также офтальмологом Франческети (Franceschetti) и в Европе известен под названием болезни или синдрома Франческети. В англо-американской литературе этот дизостоз обычно именуется болезнью или синдромом Тричер Коллинза (Treacher Collins). В мировой литературе описано около 70 случаев этого заболевания. Большинство публикаций можно найти в специальных офтальмологических, стоматологических, оториноларингологических и неврологических журналах и только в самое последнее время в рентгенологической прессе. Этот дизостоз также имеет часто семейный характер. Одинаково часто болеют мужчины и женщины. На первый план выступают абсолютно симметричные глазные симптомы, а именно весьма характерный так называемый антимонгольский разрез глаз — глазные щели направлены друг к другу под углом не кверху, а книзу. Нередко налицо колобома. Как правило, имеются различные проявления недоразвития ушей, во всяком случае наружного уха, но часто и среднего, и внутреннего уха с обеих сторон. Рентгенологически важна гипоплазия лицевых костей, главным образом челюстей, притом как верхней, с недоразвитием гайморовой полости, так и особенно нижней челюсти. Резко недоразвиты зубы, в первую очередь моляры, и выражен ряд других уже специальных стоматологических неправильностей, аномалий прикуса и т. д. Важным рентгенологическим симптомом является также отсутствие скуловых дуг, притом либо полное, либо частичное в височной части.
19. АКРООСТЕОЛИЗ
Акроостеолиз (acroosteolysis) — это врожденная болезнь, описанная впервые в 1950 г. Харнашом (Harnasch). Она наблюдается, по-видимому, не так уж редко, казуистика за 10 лет исчисляется сотней случаев. Она имеет то ярко выраженный семейный наследственный характер, то выявляется спорадически. Наблюдается она главным образом у молодых взрослых людей, у мужчин в 2—3 раза чаще, чем у женщин. Основной чертой акроостеолиза служит процесс полного костного рассасывания определенных участков скелета, в первую очередь средних отделов дистальных фаланг, с характерным сохранением как оснований, так и бугорков фаланг. Во вторую очередь литическому исчезновению подвергаются головки плюсневых костей. Процесс обычно двусторонний, но может быть не строго симметричным. Чаще всего поражаются краевые — V и I пальцы. Такие же изменения претерпевают подчас также наружные половины ключиц и альвеолярные отростки челюстей. Нередко в фалангах рук и ног, а также плюсневых и пястных костях появляются и кистовидные просветления, осевые или краевые. Остеопороза не бывает. Литический процесс лишь медленно прогрессирует. Он начинается с дистальных элементов по направлению к проксимальным. В одном нашем наблюдении акроостеолиза у 30-летней женщины (рис. 325 и 326) на одной стороне полностью рассосались кости запястья, так что пришли в соприкосновение основания пястных костей и дистальные эпифизы костей предплечья, и на той же стороне у 3-летней дочери этой больной рентгенологически обнаружено глубокое недоразвитие ядер окостенения этих же костей запястья с начинающимся укорочением лучезапястной области. Одновременно с костными изменениями проявляются и нарушения со стороны кожи и ее дериватов — передние части стоп припухают, кожа изъязвляется, притом безболезненно, размягчаются, распадаются и отторгаются ногти. К основному нервнотрофическому процессу в этих условиях может присоединиться вторичная инфекция, возникает вяло текущий остеомиелит, порой через язвы выделяются наружу костные своеобразные секвестры. Характерно также отсутствие регенеративных реакций. Потеря костных элементов неизбежно ведет к некоторому ограниченному калечению, но жизни больных акроостеолиз не угрожает (рис. 327).
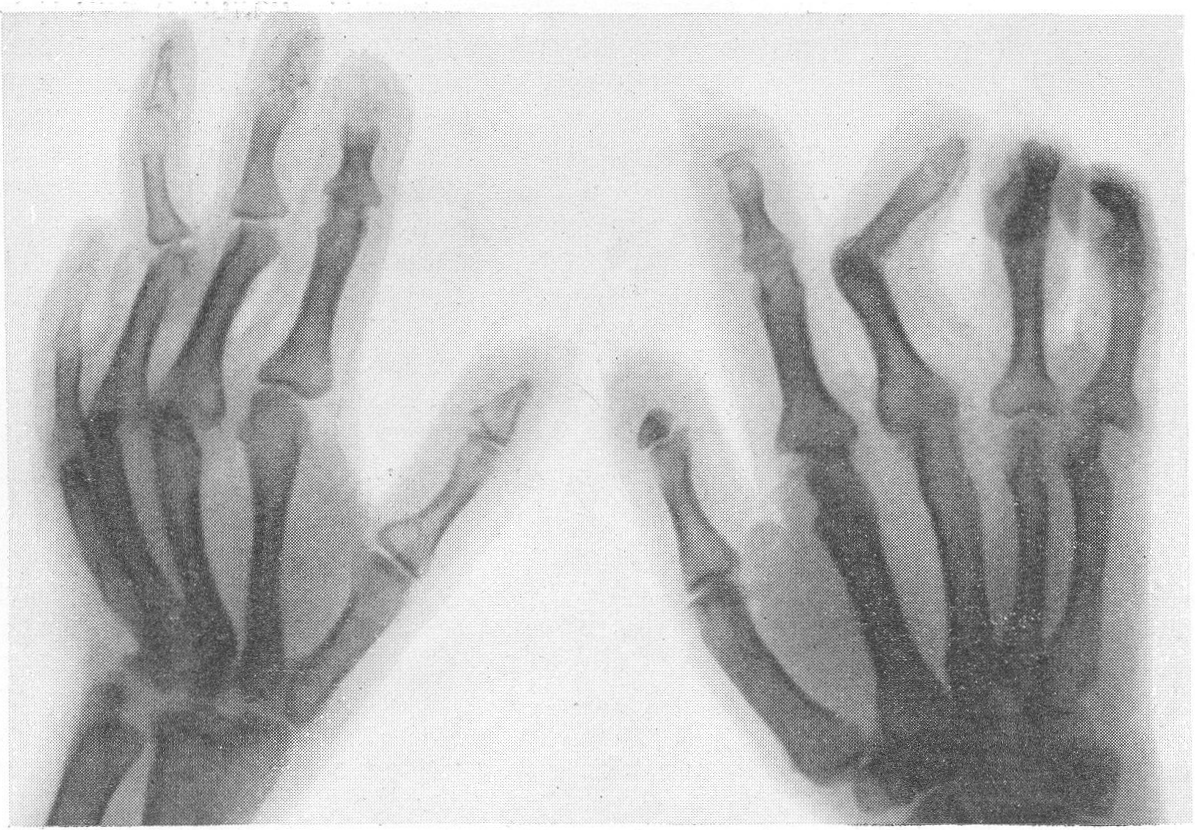{kind=link}
Рис. 325. Акроостеолиз у З0-летней женщины. Рентгенограмма обеих кистей. Полное рассасывание костей запястья слева. Остеолиз ряда ногтевых фаланг, головок основных фаланг. Деформации и мутиляции. Нарушений иннервации нет.
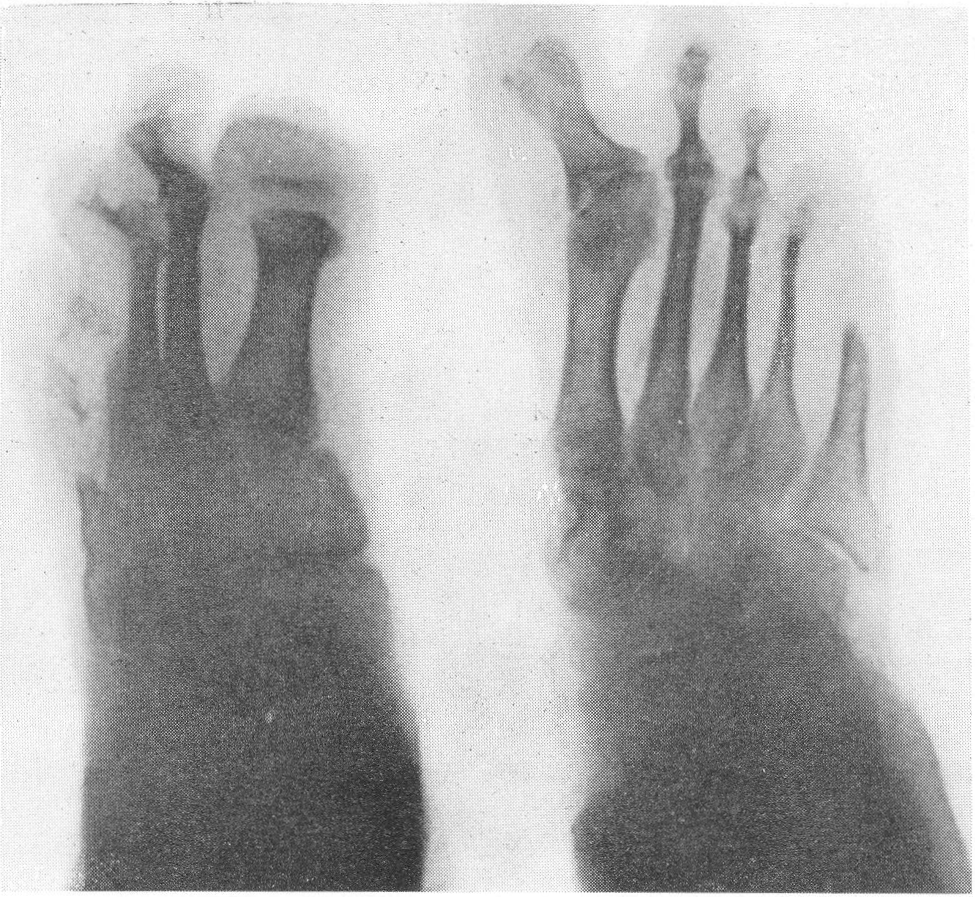{kind=link}
Рис. 326. Акростеолиз у той же больной. Рентгенограмма обеих стоп. „Подвешенные в мягких тканях” костные остатки основных фаланг большого пальца и ряда фаланг IV и V пальцев слева.
Асимметрия изменений. Патологические изменения в „акро ’’-частях тела, т. е. на периферии, могут сочетаться с различными другими аномалиями, например черепа. Рентгенологически, как правило, определяются вормиевы кости, описаны атрофические явления в отростках челюстей. Иногда большие трубчатые кости приобретают необычную-слегка изогнутую форму. Дифференциально-диагностически надо иметь в виду всю большую группу нервнотрофических болезней — поскольку поражаются главным образом нижние конечности — сухотку спинного мозга, отчасти сирингомиелию, болезнь Рейно, склеродермию, аномалии спинного мозга и всевозможные поражения нервных стволов, которые при акроостеолизе будто бы заведомо нормальны.
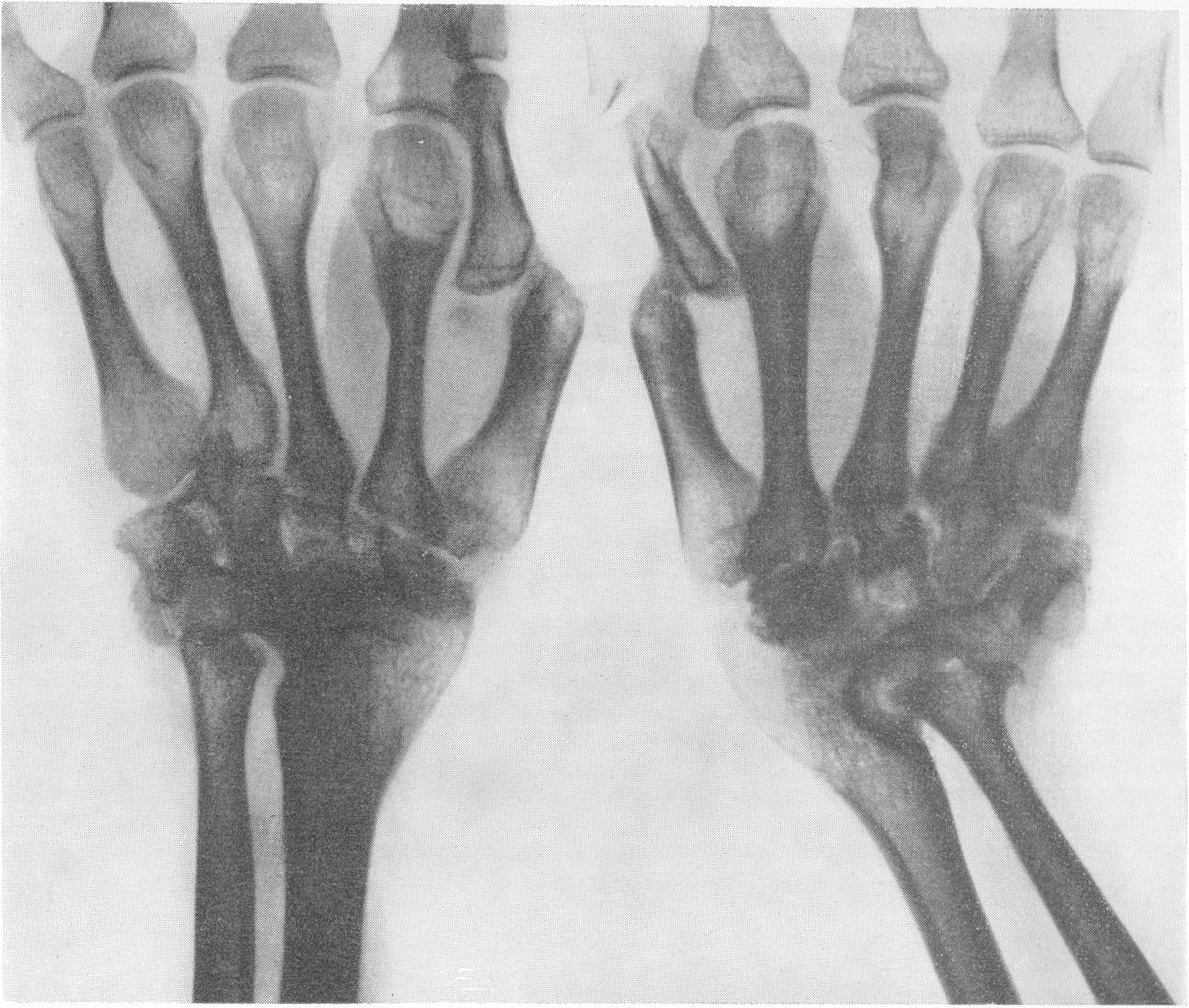{kind=link}
Рис. 327. Акроостеолиз у 40-летнего мужчины. Остеолиз костей запястья. Вывих основных фаланг большого пальца с обеих сторон. Нервная система в норме, никаких изменений чувствительной сферы.
20. АРТРОГРИПОЗ
Множественный врожденный артрогрипоз (от древнегреческих артрон — сустав и грипос — согнутый, искривленный), или врожденная амиоплазия (arthrogryposis multiplex congenita, amyoplasia congenita) — это вполне четкая нозологическая форма, редко наблюдаемое заболевание уже к моменту рождения, преимущественно у мальчиков, и в дальнейшем по существу не прогрессирующее. Название артрогрипоз предложено в 1913 г. Стерном (W. Stern). Артрогрипоз несомненно развивается уже рано в утробе матери, которая не отмечает движений ребенка. Эта болезнь выражается в сгибательных и разгибательных контрактурах ряда или многих суставов верхних и нижних конечностей, ведущих к их неподвижности, обязательно с резкой атрофией мышц. Кожа дрябла, складчата. Из-за худости мускулатуры вследствие замещения мышечной ткани фиброзной суставы кажутся относительно толстыми. Больные дети сравниваются с неподвижными деревянными куклами. Нередко налицо вторичные деформации — косолапость, вывихи и подвывихи в суставах, например вывих бедра, и другие нарушения. Череп и позвоночник остаются пощаженными. Этиология неизвестна. Рентгенологическое исследование имеет большое практическое дифференциальнодиагностическое значение. Изменения костей и суставов при артрогрипозе являются лишь вторичными. Показательна картина резкого остеопороза и атрофии костей, без каких-нибудь особых специфических для этого заболевания явлений. Большие трубчатые кости могут напомнить картину при несовершенном остеогенезе, но при артрогрипозе корковый слой не истончен. В отличительном распознавании надо иметь в виду еще врожденную амиотонию, детскую мышечную атрофию, последствия полиомиелита.
21. ГИПОФОСФАТАЗИЯ
Гипофосфатазия — это редкая хроническая, по-видимому, семейная болезнь. Ее впервые выделил в качестве очень четкой нозологической формы в 1948 г. Ретбэн (Rathbun), хотя и до этого типичные случаи данной болезни описаны в литературе в виде разрозненной и нерасшифрованной казуистики. Этиология гипофосфатазии остается пока неизвестной. В основе этой болезни лежит полное отсутствие или резкое снижение в плазме крови и в тканях щелочной фосфатазы — фермента, играющего важнейшую роль в процессе костеобразования. По этой причине у больных наступает глубокое специфическое нарушение роста и развития скелета. Гипофосфатазия поражает новорожденных, грудных младенцев и только в более редких и относительно доброкачественных случаях она, отличаясь чрезвычайно высокой смертностью, наблюдается у детей старших возрастных групп, до периода полового созревания. Нередко у одного или обоих родителей содержание фосфатазы в плазме крови также заметно понижено. Клинически на первый план выступает карликовый рост. Отдельные случаи заболевания довольно разнообразны по степени выраженности. В плазме крови определяется повышенный уровень не только кальция, но обязательно и фосфора. Гистологически кости больше всего напоминают тяжелый рахит. Обзор опубликованных в литературе 33 случаев гипофосфатазии, напечатанный в 1957 г. Куррарино с сотрудниками (Currarino), дает хорошее представление о весьма характерной рентгенологической картине этой болезни. Имеется всеобщее обеднение скелета минеральными солями. Нередко у новорожденных при наиболее тяжелых поражениях кости вообще не содержат извести, или же имеются только скудные известковые включения или центрально расположенные небольшие ядра. Иными словами, снимки производят единственное в своем роде, диагностически чрезвычайно определенное и странное впечатление — ребенок на рентгенограмме состоит как бы только из гомогенных „мягких тканей”, он как бы вообще лишен костей. В менее тяжелых случаях гипофосфатазии окостеневшими являются только в ограниченном объеме тела позвонков, диафизы длинных трубчатых костей, небольшие островки в покровных костях черепа. У выживающих старших детей бросаются в глаза исключительно широкие светлые метафизарные пространства между короткими цилиндрами окостеневших диафизов и узкими эпифизарными полосками. Большие трубчатые кости конечностей, а также ребра сильно деформированы. Нередки патологические Переломы. Как правило, имеется ранний краниостеноз.