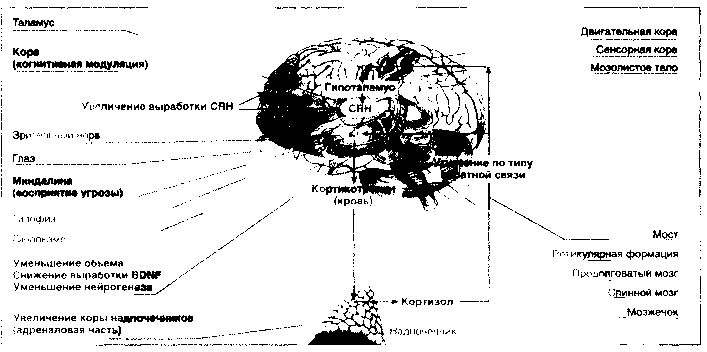
Стрессовое воздействие воспринимается корой головного мозга и передается в гипоталамус, где вырабатывается кортикотропин-высвобождающий гормон (CRH), стимулирующий гипофизарные рецепторы. Итогом этого процесса является секреция кортикотропина в плазму, стимуляция кортикотропиновых рецепторов в адреналовой области надпочечников и выброс кортизола в кровь. Воздействие на гипоталамические кортизоловые рецепторы по типу обратной связи приводит к снижению выработки CRH с целью поддержания гомеостаза (рис. 2).
В настоящее время получено достаточно много данных, свидетельствующих, что кортизол и CRH принимают участие в патогенезе депрессий. При депрессии повышено содержание кортизола в плазме, повышен уровень CRH в спинно-мозговой жидкости и увеличено содержание РНК и белка переносчика CRH в лимбических областях мозга (Merali Z., et al., 2004). Дексаметазоновый тест обнаруживает отсутствие нормальной реакции подавления выработки кортизола почти у половины больных наиболее тяжелыми формами депрессий (Carol B.J. et al., 2007). Клинический эффект тимоаналептической терапии сопровождается исчезновением положительной реакции на дексаметазоновый тест. Повышение содержания моноаминовых нейромедиаторов в синапсе также влияет на
гипоталамо-гипофизарно-адреналовую ось и приводит к уменьшению последствий длительного стресса (Holsboer Е, 2000). Возможно, что антидепрессивный эффект больше связан с коррекцией явлений вторичного стресса, вызванного болезненными депрессивными переживаниями, нежели с прямым воздействием на гипотимный аффект. Этим же механизмом можно объяснить широту клинических эффектов большинства антидепрессантов, включая лечение панического расстройства, посттравматического стрессового расстройства, булимии, предменструального синдрома и обсессивно-компульсивного расстройства.
{kind=link}
Примечание.
Гипоталамо-гипофизарно-кортизоловая гипотеза депрессии постулирует, что в ее основе лежит нарушение кортизоловой реакции в ответ на стрессовое воздействие. На схеме стрелками указано, что на стресс реагируют прежде всего кора и миндалина мозга, откуда сигнал передается в гипоталамус, где происходит выработка кортикотропинвысвобождающего гормона (CRH), побуждающего секрецию кортико-тропина в передней части гипофиза, который поступает непосредственно в кровь и стимулирует адреналовую часть коры надпочечников и соответствующую секрецию глюкокортикоидного гормона — кортизола. Кортизол по механизму обратной связи тормозит продукцию CRH и кортикотропина соответственно в гипоталамусе и в гипофизе. Исследования, поддерживающие гипоталамо-гипофизарно-коргизоловую гипотезу депрессии, включают: повышение уровня кортизола в крови при тяжелых депрессиях; увеличение объема передней части гипофиза и адреналовой коры надпочечников; повышение содержания CRH в церебро-спинальной жидкости; усиление экспрессии CRH в лимбических структурах; уменьшение объема гиппокампа, числа нейронов и глиальных клеток, что, по-видимому, связано с угнетением нейрогенеза, вызванного повышенным содержанием кортизола и снижением выработки мозгового нейротрофического фактора (BDNF).
Антагонисты CRH-рецепторов показали обнадеживающие результаты в экспериментах у животных, однако не обнаружили отчетливой антидепрессивной активности в клинических условиях (Louis С., et al, 2006; Kehne J. H., 2007; Binneman В. et al., 2008). При тяжелых психотических депрессиях был зарегистрирован эффект блокаторов глюкокортикоидных рецепторов, частности мифепристона (Flores В. Н. et al., 2006; De Battista C. et al., 2006; Beasey С. M. et al., 2009). Мифепристон также усиливает нейрогенез (Mayer J.N. et al., 2006) и повышает чувствительность 5-HT2A рецепторов в экспериментальных моделях (Trajkovska V. et al., 2009).
Важное значение в патогенезе депрессий, по-видимому, имеют патологические циркадианные колебания содержания кортизола в крови, особенно длительное повышение уровней кортизола в ночное время, когда у здоровых лиц он практически отсутствует. Возможно также, что периферический подъем кортизола в крови лишь отражает центральные нарушения в системе CRH и его патологическую реакцию на длительное стрессовое воздействие.
Повышенное содержание глюкокортикоидов снижает нейропластичность мозга, включая способность к нейрогенезу. При депрессии уменьшается объем гиппокампа (MacQueen G. М. et al., 2003). В посмертных исследованиях у больных депрессией были обнаружены потеря клеток в поясничной извилине префронтальной коры, атрофические изменения в дорсолатеральной префронтальной и орбитофронтальной коре, а также увеличение числа нервных клеток в гипоталамусе и дорсальной части ядра шва (центральная часть серотонинергической системы мозга) (Rajkowska G., 2000; Koolschijn Р. С. et al., 2009). Эти находки похожи на артофические изменения в мозге при болезни Кушинга, а также у экспериментальных животных, находившихся под воздействием больших доз глюкокортикоидов. Нужно отметить, однако, что при депрессии подъем кортизола в крови значительно меньше, чем при болезни Кушинга. Стресс подавляет нейрогенез у животных, включая приматов, а антидепрессанты могут предотвращать подавление нейрогенеза в этих моделях и даже стимулируют нейрогенез (Porera Т. D. et al., 2007). Клиническое значение этого феномена у больных депрессией остается неясным.
Мозговой нейротрофический фактор (BDNF) является главным нейротрофическим пептидом в организме, ответственным за процессы нейропластичности, включая рост аксонов, увеличение числа синапсов и выживания клеток. Содержание BDNF снижается под воздействием стресса и кортизола (Angelucci Е et al., 2005; Kozlovsky N. et al., 2007). Посмертные исследования мозга у жертв суицидов, страдавших депрессией, обнаружили снижение концентрации BDNF в гиппокампе (Karege F. et al., 2005). Антидепрессанты и ЭСТ увеличивают выработку BDNF и гормона роста (Chen В. et al., 2001). В одном из исследований было показано уменьшение объема гиппокампа у больных депрессией с аллелем BDNF по metl66 (Frodl Т. et al., 2007). Не исключено, что BDNF является связующим звеном между стрессом, нейрогенезом и атрофией гиппокампа при депрессии. Вместе с тем, генетические исследования полиморфизма гена BDNF по vaIl66 met не обнаружили связи с депрессией. По-видимому, BDNF является относительно неспецифическим механизмом, задействованным при различных психических заболеваниях. Блокирование гена BDNF у мышей не вызывает депрессоподобной реакции (Lyons W. Е. et al., 1999), однако может снижать эффект антидепрессантов (Duman R. S., Monteggia L. М.,2006). Воспалительные процессы и некоторые нейротоксины также увеличивают содержание BDNF в мозге (de Pablos R. М. et al., 2006). Хотя антидепрессанты самостоятельно могут вызывать образование новых синапсов, способствуя более эффективной переработке внешних импульсов (Castren Е., 2005), большинство из них меняют экспрессию BDNF (Watanabe К. et al., 2010).
При стрессовой реакции посредством нейропептидов усиливается глутаматная нейротрансмиссия, что было продемонстрировано при депрессиях (Zarate С. A. et al., 2004; Feyssa А. М. et al., 2009). Выброс глутамата (ГЛУ) и возбуждающих нейропептидов увеличивает нейротоксичность и ведет к преждевременному клеточному апоптозу. Антагонисты NMDA рецепторов (кетамин, рилузол и траксопридил) дают быстрый, но кратковременный антидепрессивный эффект при терапевтически резистентных депрессиях (Sanacora G. et al., 2004; Zarate C. A. et al., 2006; Preskorn S. H. et el., 2008; Phepls L. E. et al.,2009). Недавно способность к нормализации глутаматергической трансмиссии была обнаружена у известного антидепрессанта с недостаточно понятным механизмом действия тианептина (Mс Ewen В. S. et al., 2010).
Эпидемиологические данные указывают на то, что депрессия часто предшествует сердечно-сосудистым заболеваниям, осложняет их течение и приводит к повышенной смертности (Lesperance F et al., 2007). В патогенезе как депрессии, так и сердечно-сосудистой патологии участвует ряд общих факторов, в частности, недостаток омега 3 жирных кислот (Nemets В. et al., 2002) и увеличение содержания в плазме гомоцистеина (Folstein М. et al., 2007). Повышенный уровень кортизола в крови при депрессии увеличивает риск поражения коронарных сосудов, поскольку кортизол способствует развитию висцерального ожирения (Brown Е. S. et al., 2004). Применение антидепрессантов увеличивает выживание больных с инфарктом миокарда (Taylor С. В. et al., 2005). При депрессии и сердечно-сосудистых заболеваниях описываются признаки воспалительного процесса (Muller N., 2006). Возможно, что в патогенезе депрессии и сердечно-сосудистых заболеваний важную роль играет поражение эндотелиальных клеток, которые продуцируют BDNF и запускают процессы нейрогенеза.
Не менее важную роль в патогенезе депрессии, по-видимому, играет и нарушение иммунной функции (Miller А. Н. et al., 2009). При депрессии значительно повышен уровень провоспалительных цитокининов, таких как интерлейкин6 (IL-6) и α-фактор некроза опухолей (TNFα), который снижается у больных с эффектом антидепрессантов (Capuron L. et al., 2003; O’Brien S. М. et al., 2007; Hernandez M. E. et al., 2008). Содержание этих цитокининов повышено при многих соматических заболеваниях, включая рак, и коррелирует с развитием депрессии при этих состояниях. Введение этих факторов животным вызывает психомоторную заторможенность, снижение сексуальной активности и ангедонию (Anisman Н. et al., 2005; Janicki-Deverts D. et al., 2007). Несколько специфических лекарственных средств — инфликсимаб (TNFα ингибитор) и целекоксиб (СОХ-2 ингибитор), в предварительных исследованиях обнаружили антидепрессивную активность (Akhondzaden S. et al., 2009; Muller N„ 2010).
Стрессорные гормоны, моноамины и нейротрофические факторы могут влиять на циркадианные ритмы, приводя к развитию депрессии (Burke Н. М. et al., 2005; Ramirez-Rodriguez G. et al., 2009).